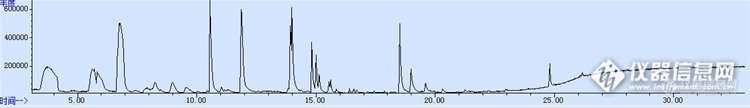
之前做顶空固相微萃取气质联用还没有那么多杂峰,如图1.后来仪器关机再开机,就出现很多杂峰,如图2,约3.6min,6.6min,10.442min,19min等几个峰都是杂峰,大多数硅氧烷化合物,走空针的时候没有杂峰,我换了进样口的隔垫还是这样。求大神帮忙!http://ng1.17img.cn/bbsfiles/images/2016/12/201612191807_01_3160951_3.jpghttp://ng1.17img.cn/bbsfiles/images/2016/12/201612191807_02_3160951_3.jpg
二氧杂芑 二氧杂芑是科学上所熟知的一种毒性最强的化学物类别,美国环保署(EPA)於1994年9月所发现的一篇供大众述评的草拟报告就把二氧杂芑称为最严重威胁人体健康之物,二氧杂芑对公众健康之影响足以比得上1960年代DD对公众健康所带来的影响作用。二氧染芑是否会致癌?二氧染芑的确会致癌,根据EPA的报告,证实二氧染芑是癌症的危险之源,1997年2月,隶属世界卫生组织的国际癌症研究机构(IARC)公然宣布,最”"强效"的二氧杂芑是一级致癌物,意指一种"已知的人体致癌物",2002年7月,一项研究结果证实,二氧杂芑与不断增加的乳癌罹患率有密切关系,因长期接触二氧杂芑,而引致的其他相关健康问题有哪些?因长期接触二氧杂芑,而引致的其他相关健康问题有哪些。除了致癌之外,长期接触二氧杂芑也会造成严重的生育和发育问题,二氧杂芑会对人体的免疫系统和激素系统造成破坏及干扰,而所引致的相关问题包括畸胎、小产、生育能力减退、精子量减少、子宫内膜异位、糖尿病、无学习能力、免疫系统阻滞、肺部问题、皮肤病、睾丸素分泌量降减等等。你是否应该关注二氧杂芑所造成的威胁?当然应有所警觉,并确定鱼油产品经被证实不含二氧杂芑。根据EPA的相关报告中,二氧杂芑被称为亲脂物质,意思是,在被二氧杂芑污染的渔场地区,二氧杂芑会迅速积聚在鱼群体的,而不会继续停留於水中,这些有毒的化学物质经过食物链在鱼群体的累积,而二氧杂芑量比四周环境中所存再的二氧杂芑量多达十万倍。
有第三方可以测试五氧化铌金属杂质的单位吗?急需!!QQ 346805152

寒冬来临,暖暖的一碗羊杂或者牛杂汤,浇上红红的辣椒油,热热乎乎的,让你吃到浑身舒坦!http://ng1.17img.cn/bbsfiles/images/2014/12/201412072155_526159_1609327_3.jpg可是,很多人不喜欢吃这些东西。原因就是,所谓羊杂、牛杂,就是羊下水、牛下水,就是羊或牛的内脏,包括肝、肺、肚、肠等...这些东西吃了,对人体会有好处吗?
如题,俺第一次测盐酸左氧氟沙星,做有关物质时杂质A与左氧保留时间完全重叠,排除了乙酸铵、高氯酸钠等试剂滴原因,实在没辙咧,请教大虾帮忙。盐酸左氧氟沙星有关物质测定方法(来源:中国药典2010年版第一增补本): 有关物质 取本品,精密称定,加0.lmol/L盐酸溶液溶解并定量稀释制成每1ml中约含1.2mg的溶液,作为供试品溶液,精密量取适量,用0.1mol/L盐酸溶液定量稀释制成每1ml中含2.4ug的溶液,作为对照溶液。另精密称取杂质A对照品约18mg,置100ml量瓶中,加6mol/L氨溶液1ml与水适量使溶解,用水稀释至刻度,摇匀,精密量取2ml,置100ml量瓶中,用水稀释至刻度,摇匀,作为杂质A对照品溶液。照高效液相色谱法(附录V D)测定,用十八烷基硅烷键合硅胶为填充剂;以醋酸铵高氯酸钠溶液(取醋酸铵4.0g和高氯酸钠7.0g,加水1300ml使溶解,用磷酸调节pH值至2.2)-乙腈(85 :15)为流动相A,乙腈为流动相B;按下表进行线性梯度洗脱。柱温为40°C;流速为每分钟1ml。称取左氧氟沙星对照品、环丙沙星对照品和杂质E对照品各适量,加0.1mol/L盐酸溶液溶解并稀释制成每1ml中约含左氧氟沙星1.2mg、环丙沙星和杂质E各6ug的混合溶液,取10ul注人液相色谱仪,以294nm为检测波长,记录色谱图,左氧氟沙星峰的保留时间约为15分钟。左氧氟沙星峰与杂质E峰和左氧氟沙星峰与环丙沙星峰的分离度应分别大于2.0与2.5。量取对照溶液10ul注人液相色谱仪,以294mn为检测波长,调节检测灵敏度,使主成分色谱峰的峰高约为满量程的20%。精密量取供试品溶液、对照溶液和杂质A对照品溶液各10ul,分别注人液相色谱仪,以294nm和238nm为检测波长,记录色谱图。供试品溶液色谱图中如有杂质峰,杂质A(238nm检测)按外标法以峰面积计算,不得过0.3%。其他单个杂质(294nm检测)峰面积不得大于对照溶液主峰面积(0.2%),其他各杂质(294nm检测)峰面积的和不得大于对照溶液主峰面积的2.5倍(0.5%)。供试品溶液色谱图中任何小于对照溶液主峰面积0.1倍的峰可忽略不计。时间(分钟) 流动相A(%) 流动相B(%) 0 100 0 18 100 0 25 70 30 39 70 30 40 100 0 50 100 0
我们有一种氧化铝粉末状的物质,是做蓝宝石的原材料,想检测氧化铝的纯度,以及一些微量杂质的含量,具体的杂质含量参见附件。我希望可以找个可以对外测试的机构,我在深圳地区。
最近强生婴儿用品有毒事件很火,据检测报告显示甲醛和1,4-二氧杂环乙烷导致过敏,哪位大虾可以提供关于1,4-二氧杂环乙烷的信息?包括CAS号,结构式等,谢谢!E-mail: ljmw521@163.com
想请教一下各位老师GC色谱上的某峰我怀疑是杂质A,它保留时间和A的是一样的,往里面加A这个杂质峰面积确实变大了(回收率是98%),能不能证明这个峰就是A的峰?还是说有可能是其他极性相似的杂质?类推一下,想问问加样实验之后增加了的那个峰一定是你加进去的那个杂质的吗?(在确定已经含有该成分之后)A是个酸,之前走其他柱子可以检出主成分的其他杂质,换了极性柱之后只剩下主峰和疑似A的峰检测器是FID
氯化镁测试氧化镁杂质的方法?求教,最好有标准
首先祝大家圣诞快乐、新年快乐![em24] 我向大家请教的问题是如何用HRTEM表征掺杂纳米氧化物半导体。基体为10nm左右的纳米氧化物,掺杂相为稀土离子,在基体内分布比较均匀,但是无定形的。做HRTEM的目的在于想确定无定形掺杂相在基体中的位置,是间隙掺杂还是取代掺杂。另外,用哪种HRTEM较好,LaB6 HRTEM、FEG HRTEM 还是STEM? 欢迎大家不吝赐教!谢谢先!
如下图,柱子老化后走了样品后目标物周围存在杂峰无法分离,后走标样,标样周围也出现两个杂峰,请大家帮忙看看是什么原因造成的,是柱子污染?标样的问题?或者其他原因,可以怎么去验证,谢谢 [img]https://ng1.17img.cn/bbsfiles/images/2020/03/202003192328253206_6217_4104781_3.png[/img]
请问各位前辈,现在要用ICP检测氧化锌、氧化镍、四氧化三钴中杂质金属元素的测试,请问选线时选信噪比高和灵敏度高的就行吗?谢谢!

红外光谱求助, 下图是纯氧化锡及Ni掺杂氧化锡的红外光谱,最下面的线是纯氧化锡,其他是不同含量的Ni掺杂氧化锡,主要是1441这个峰没办法解释啊。XRD 显示主要物质是氧化锡和氧化镍 可能还有其他锡或者镍的物质 1441的这个峰没有办法解释啊 他是Ni掺杂后的出现的 峰强也很高 感觉是跟Ni有关系的 但是没办法解释这是什么峰啊[img=,750,530]http://ng1.17img.cn/bbsfiles/images/2017/06/201706050945_01_3238542_3.jpg[/img]----将图片直接放到帖子中了。PS:已转到 IR版。下次发帖记得到对应的技术版面,新手版面的浏览量小,可能会耽误您的问题。
请问各位高手,如何确定银金属中含有氧或其它杂质?[em34] [em34] [em49]
那位大侠知道[url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]分析工业用环氧丙烷中杂质的方法?急。。
各位老师帮忙看下怎么处理,谢谢了!做的是归脾丸,流动相是甲醇:水(75:25)用的是高纯氮气,0.2Mpa温度设置T2:110 T1:60对照有小杂峰不多,进样后出现好多小杂峰是什么情况,请各位老师帮忙看下谢谢了!
求助:给位大侠,有没有做过氧氟沙星氯化钠注射液的??我一直弄不明白2010药典对杂质的计算,各位如果做过,麻烦发一张积分图谱,如果有数据报告最好,谢谢注:双波长的转换我会做,就是不知道哪个是我所需要的峰?
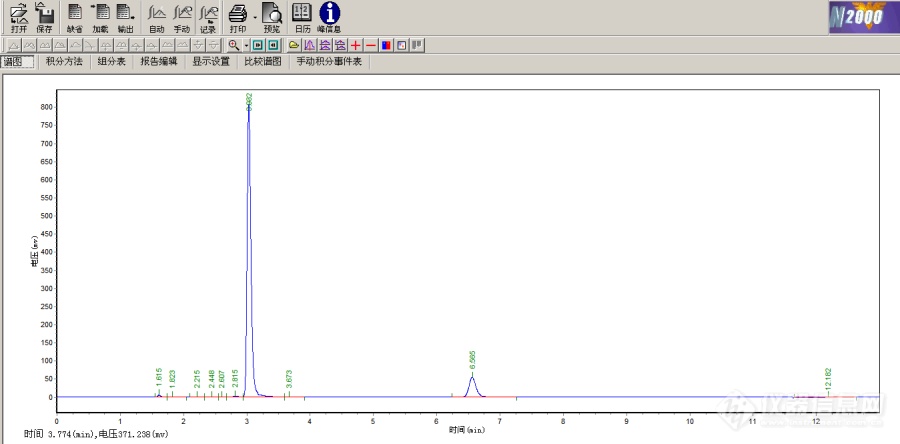
[img=吡虫啉+戊唑醇标样谱图,690,340]https://ng1.17img.cn/bbsfiles/images/2022/06/202206081600415707_2074_5607689_3.png!w690x340.jpg[/img][img=吡虫啉标样峰谱图放大,690,292]https://ng1.17img.cn/bbsfiles/images/2022/06/202206081601249731_4469_5607689_3.png!w690x292.jpg[/img]吡虫啉戊唑醇的标样峰谱图,吡虫啉的标样峰感觉是有杂质还是机器的问题色谱柱:Mars120 C18 5.0微米,250nmX4.6nm
如题~最近使用mCPBA环氧化烯烃时,用二氯甲烷溶解,冰浴反应5min,室温放置10min后,加10%亚硫酸钠溶液洗涤离心,除去过量间氯过氧苯甲酸后,再用10%碳酸钠溶液洗涤,离心,再水洗,氮吹后上机分析,结果不太如意,进GC-FID分析后出现了更多杂质,mCPBA纯度70%,不知道问题出在哪,有没有做过此反应的大神,求指导!!!
气相顶空进样空白也有杂峰怎么回事,而且几分钟就开始基线不稳?已经老化了,而且也加压吹扫了很久还是这样!
请问各位专家: 对甲氧苯甲酰氯中的杂质限量及分析方法。 谢谢!
安捷伦6820是咋样确定分流比以及咋样确定隔垫吹扫流量的?
如何应用ICP分析高纯氧化铝中的杂质元素
情况说明:1、甲氰菊酯和联苯菊酯为同一批次配制。容量瓶用洗洁精洗过,怀疑是洗洁精杂峰,导致标样污染配制错误。2、氟氰戊菊酯为今天配制。为新的容量瓶,新的移液管,用纯水超声波后正已烷润洗3次,排除配制原因及洗浩精原因。3、出现相同杂峰后怀疑正已烷试剂污染,用正已烷(旧)(直接从瓶中取样)进样,没有杂峰。4、正已烷(旧)进样后没有杂峰,怀疑是否是容量瓶被污染,把正已烷转移到容量瓶,摇匀旋涡,充分混匀后取正已烷(新)进样,没有杂峰。5、进空针,排除色谱柱,检测器污染。 以上全部用ECD检测。ECD经过昨天一晚上300度烘烤,平衡基线。6、进亚胺硫磷(FPD)没有杂峰,初步排除色谱柱污染。7、仪器为新仪器,安装时走过一针ECD,色谱图没有杂峰。后一直放置3个月,近一个星期开始使用,色谱柱为HP-5已老化。衬管为新衬管。现在我做的初步情况就是这样,请大家帮忙分析一下是哪个地方出现原因。
安捷伦6820是咋样确定分流比以及咋样确定隔垫吹扫流量的?
各位老师,我最近收到一个样品,氧化镁掺杂3%的磷酸,TG-DSC支架,加热到1100 oC会污染吗?氧化镁,我觉得没有啥问题,磷酸分解是不是生成P2O5,好像P2O5在300多度就升华的。是不是很危险呢?
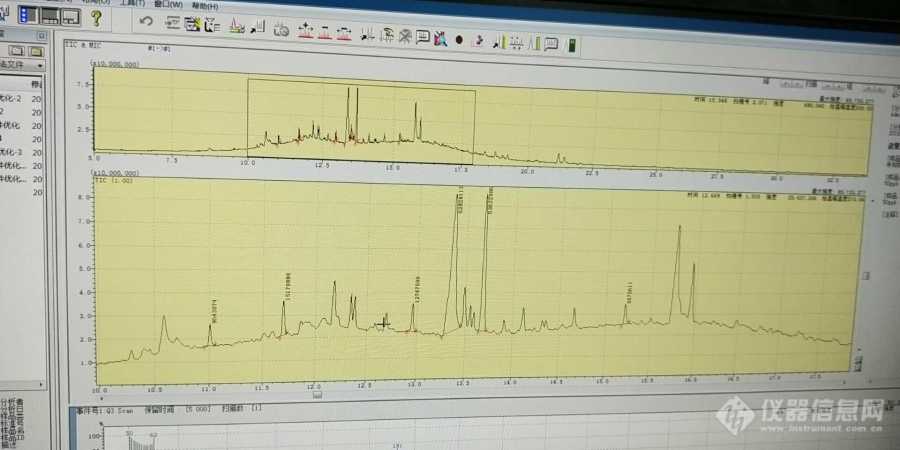
最近在用gcms做实验,测得是有机氯,采用不分流进样,之前还是很好没有杂峰,但是最近测样时出现很多杂峰,已经老化过色谱柱两次了,也换了衬管和进样隔垫,但是没用,希望大家可以帮帮忙,谢谢!
各位童鞋,哪里有出售三水氧化铝杂晶相含量测定用标准样品?麻烦告诉一下,谢谢了!
需要测试氢氧化铝中的杂质含量,但是很难溶解,有没有谁有好的办法。硝酸,盐酸,硫酸等,拜托!
最近天天发贴求助,就是色谱进样后有时(大部分时间)不出峰,有时候发现灭火,有时候发现进样针堵了,(不知道这些是否是真正原因,这些问题解决后),这些问题解决后接下来就出现长时间大量杂质峰,在检测条件柱温200度,进样200度,辅助箱200度,检测器200度得走三个多小时,其中杂质峰密密麻麻,下一针后还是这样,我的样品是自己配的不可能那么多杂质,这是什么原因呢,期待中~~~







 我要推广仪器
我要推广仪器
 下载APP
下载APP