基于镜像酶正交酶切的蛋白质复合物规模化精准分析新方法
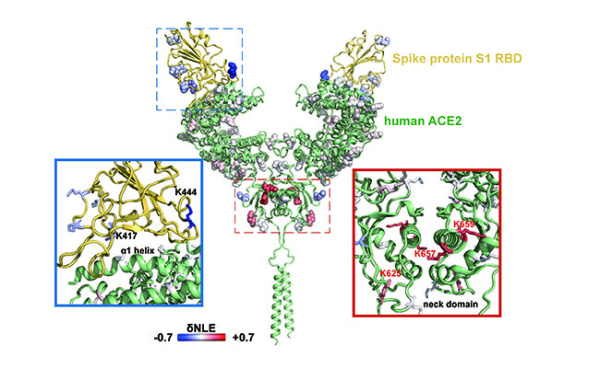
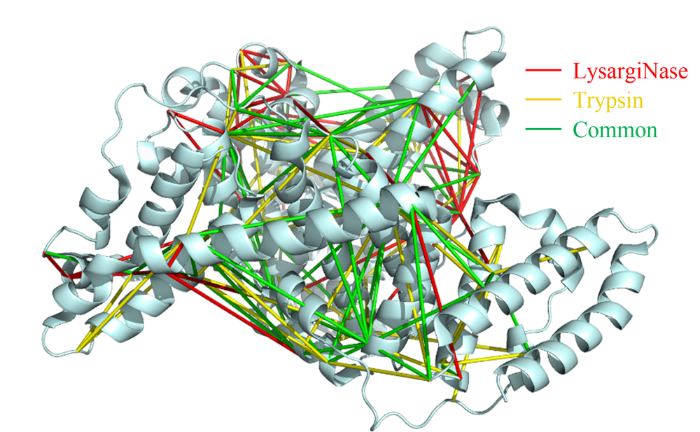
蛋白质作为生命活动的执行者,通过自身结构的动态改变,以及与其他蛋白质相互作用组装为蛋白质复合物,调控各种生物学过程。因此,如何实现蛋白质复合物的精准解析已成为当前生命科学的研究热点。化学交联结合质谱(CXMS)技术作为蛋白质复合物解析的新兴技术,利用化学交联剂将空间距离足够接近的蛋白质分子内或分子间的氨基酸残基以共价键连接起来,再利用液相色谱-质谱联用对交联肽段进行鉴定,实现蛋白质复合物的组成、界面和相互作用位点的解析。该技术具有分析通量高、灵敏度高、可提供蛋白质间相互作用的界面信息、普遍适用于不同种类和复杂程度的生物样品等优势,已成为X射线晶体衍射、低温冷冻电镜、免疫共沉淀等蛋白质复合物研究技术的重要补充。化学交联位点的鉴定覆盖度和准确度决定着该技术对于蛋白质复合物结构的解析能力。目前,为了实现蛋白质复合物的高覆盖度交联,研究人员发展了可用于共价交联赖氨酸(K)的氨基、谷氨酸(E)/天冬氨酸(N)的羧基、精氨酸(R)的胍基以及半胱氨酸(C)的巯基等多种活性基团的新型交联剂。进而,为了提高低丰度交联肽段的鉴定灵敏度,体积排阻色谱法、强阳离子交换色谱法,及亲和基团富集策略被提出用于交联肽段的高选择性富集,如可富集型化学可断裂交联剂——Leiker,与不具备富集功能的交联剂相比,通过亲和富集可以将交联位点鉴定数目提高4倍以上。胰蛋白酶镜像酶(LysargiNase)的酶切位点与胰蛋白酶互为镜像,可特异地切割赖氨酸和精氨酸的N端。由于LysargiNase的N端酶切特点,电荷主要分布在交联肽段的N端,在碰撞诱导裂解(CID)和高能诱导裂解(HCD)模式下产生以b离子为主的碎片离子,与胰蛋白酶酶切肽段以y离子为主的碎片离子互为镜像补充,为胰蛋白酶酶解肽段在质谱鉴定中b离子缺失严重的问题提供了很好的解决办法。由于具有较高的酶切特异性和酶活性,镜像酶已经成功地应用于蛋白质C末端蛋白质组鉴定、磷酸化蛋白质组研究、甲基化蛋白质组鉴定等方面,然而在CXMS中的应用仍未见报道。为进一步提高对蛋白质复合物结构及相互作用位点的解析能力,本文发展了LysargiNase与胰蛋白酶联合酶切的方法,基于镜像酶正交切割的互补特性,通过产生赖氨酸及精氨酸镜像分布的交联肽段,以增加特征碎片离子数量和肽段匹配连续性,从而提升交联肽段的谱图鉴定质量,达到提高交联位点的鉴定覆盖度和准确度的目的。通过分别对牛血清白蛋白及大肠杆菌全蛋白样品的交联位点鉴定结果的考察,评价该策略对单一蛋白样品和复杂细胞裂解液样品蛋白质复合物表征的应用潜力。蛋白质样品制备称取牛血清白蛋白粉末,以20 mmol/L 4-(2-羟乙基)-1-哌嗪乙磺酸(HEPES, pH 7.5)作为缓冲体系,配制0.1 mmol/L牛血清白蛋白溶液。大肠杆菌细胞(种属K12)在37 ℃下采用Luria-Bertani(LB)培养基培养24 h,然后于4 ℃以4000 g离心2 min,收集细胞沉淀。细胞沉淀采用磷酸盐缓冲液(PBS)清洗3遍后,悬浮于细胞裂解液(含20 mmol/L HEPES和1%(v/v)蛋白酶抑制剂)中,冰浴超声破碎180 s(30%能量,10 s开,10 s关)。匀浆液于4 ℃以20000 g离心40 min,收集上清,采用BCA试剂盒测定所得蛋白质含量。稀释大肠杆菌蛋白裂解液至蛋白质含量为0.5 mg/mL。化学交联样品制备以20 mmol/L HEPES(pH 7.5)为溶剂配制浓度为20 mmol/L 的BS3交联剂母液 将交联剂母液加入牛血清白蛋白的缓冲溶液及大肠杆菌蛋白裂解液中,使交联剂的终浓度为1 mmol/L,在室温条件下反应15 min 通过添加终浓度为50 mmol/L的淬灭溶液NH4HCO3进行交联反应淬灭,并在室温下孵育15 min 在冰浴条件下,将交联样品逐渐滴入8倍体积的预冷丙酮中,于-20 ℃静置过夜 在4 ℃条件下,以16000 g转速离心,去除丙酮,然后将交联蛋白用预冷丙酮清洗2次,去除上清液后,于室温挥发掉残余的丙酮 以8 mol/L尿素溶液复溶蛋白质沉淀 将牛血清白蛋白交联样品以5 mmol/LTCEP作为还原剂,于25 ℃下反应1 h进行变性和还原 将大肠杆菌样品以5 mmol/LDTT作为还原剂,于25 ℃下反应1 h进行变性和还原,避免大肠杆菌蛋白在酸性条件下发生变性 添加终浓度为10 mmol/L的碘乙酰胺(IAA),在黑暗中,于室温下反应30 min 以50 mmol/LNH4HCO3稀释样品至尿素浓度为0.8 mol/L后,将样品均分为两份,一份以蛋白样品与蛋白酶的质量比呈50:1的比例加入胰蛋白酶,于37 ℃酶解过夜,另一份加入终浓度为20 mmol/L的CaCl2,以蛋白样品与蛋白酶的质量比呈20:1的比例加入LysargiNase,并在37 ℃温度下酶解过夜。液相色谱-质谱鉴定及数据搜索上述所有样品经过除盐,使用0.1%甲酸(FA)溶液复溶,用超微量分光光度计测定肽段浓度,进行反相高效色谱分离和质谱分析。牛血清白蛋白样品采用Easy-nano LC 1000系统偶联Q-Exactive质谱仪平台进行质谱分析。流动相A: 2%(v/v)乙腈水溶液(含0.1%(v/v)FA) 流动相B: 98%(v/v)乙腈水溶液(含0.1%(v/v)FA)。梯度洗脱程序:0~10 min, 2%B~7%B 10~60 min, 7%B~23%B 60~80 min, 23%B~40%B 80~82 min, 40%B~80%B 82~95 min, 80%B。Q-Exactive质谱仪采用数据依赖性模式,Full MS扫描在Orbitrap上实现,扫描范围为m/z 300~1800,分辨率为70000(m/z=200),自动增益控制(AGC)为3×106,最大注入时间(IT)为60 ms,母离子分离窗口为m/z 2。MS/MS扫描的分辨率为17500(m/z=200),碎裂模式为HCD,归一化碰撞能量(NCE)为35%, MS2从m/z 110开始采集,MS2的AGC为5×104, IT为60 ms,仅选择电荷值为3~7且强度高于1000的母离子进行碎裂,并将动态排除时间设置为20 s。每个样品分析3遍。大肠杆菌样品采用EASY-nano LC 1200系统偶联Orbitrap Fusion Lumos三合一质谱仪平台进行质谱分析。流动相A: 0.1%(v/v)甲酸水溶液 流动相B: 80%(v/v)乙腈水溶液(含0.1%(v/v)FA)。梯度洗脱程序:0~28 min, 5%B~16%B 28~58 min, 16%B~34%B 58~77 min, 34%B~48%B 77~78 min, 48%B~95%B 78~85 min, 95%B。Orbitrap Fusion Lumos三合一质谱仪采用数据依赖性模式,Full MS扫描在Orbitrap上实现,扫描范围为m/z 350~1500,分辨率为60000(m/z=200), AGC为4×105, IT为50 ms,母离子分离窗口为m/z 1.6。MS2扫描的分辨率为15000(m/z=200),碎裂模式为HCD, NCE为30%, MS2从m/z 110开始采集,MS2的AGC为5×104, IT为60 ms。仅选择电荷值为3~7且强度高于2×104的母离子进行碎裂,并将动态排除时间设置为20 s。每个样品分析3遍。质谱数据文件(*.raw)采用pLink 2软件(2.3.9)对交联信息进行检索和鉴定。使用从UniProt于2019年4月27日下载的牛血清白蛋白序列和大肠杆菌序列,搜索参数如下:酶切方式为胰蛋白酶(酶切位置:K/R的C端)、LysargiNase(酶切位置:K/R的N端) 漏切位点个数为3 一级扫描容忍(precursor tolerance)2.00×10-5 二级扫描容忍(fragment tolerance)2.00×10-5 每条肽段的质量范围为500~1000 Da 肽段长度的范围为5~100个氨基酸 固定修饰为半胱氨酸还原烷基化(carbamidomethyl [C]) 可变修饰为甲硫氨酸氧化(oxidation [M])、蛋白质N端乙酰化(acetyl [protein N-term]) 肽段谱图匹配错误发现率(FDR)≤5%。映射胰蛋白酶与LysargiNase酶解样品的交联位点在牛血清 白蛋白晶体结构(PDB: 3V03)的映射 LysargiNase与胰蛋白酶酶解样品的交联位点对及单一交联位点的互补性LysargiNase与胰蛋白酶酶解样品共同得到的交联位点鉴定打分比较b+/++与y+/++离子碎片分别在α/β-肽段的碎片覆盖度LysargiNase与胰蛋白酶酶解的交联肽段质谱图大肠杆菌样品中LysargiNase与胰蛋白酶酶切鉴定蛋白质复合物信息互补性带点击了解原文:https://www.chrom-china.com/article/2022/1000-8713/1000-8713-40-3-224.shtml






















 我要推广仪器
我要推广仪器
 下载APP
下载APP