高内涵——基于FRET分析活细胞中的ERK信号转导
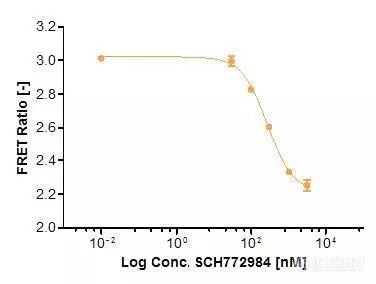
Extracellular signal-regulated kinase(ERK)是胚胎发生,细胞分化,细胞增殖和细胞死亡调控的关键组成部分。ERK途径起源于质膜中的活化受体,并通过Ras/Raf/MEK至ERK(图1)。图1. Ras/Raf/MEK/ERK信号级联将信号从细胞表面受体如EGF受体(EGFR)传播到细胞内蛋白质。ERK是该途径的最终组分,并且在被生长因子(例如EGF(表皮生长因子))激活后,触发下游效应,如激酶或转录因子的激活。该途径被不同类型的受体激活,包括受体酪氨酸激酶 (例如EGF受体)以及G蛋白偶联受体。作为信号传导途径的最终组分,ERK磷酸化不同的细胞内蛋白质,包括大量其他激酶和转录因子。ERK信号传导途径存在于各种癌症类型中,因此正在研究作为治疗干预的靶标。在这里,我们描述了如何在Operetta CLS高内涵分析系统上自动化研究ERK信号传导的活细胞FRET测定。该测定可以用于药物发现。基于FRET的ERK生物传感器FRET是从供体分子到受体分子的非辐射能量转移。能量转移需要供体和受体间隔小于10nm,因此提供了研究分子接近度变化的敏感工具,例如蛋白质 - 蛋白质相互作用(分子间FRET)或蛋白质的构象变化(分子内FRET)。在这项研究中,我们专注于分子内FRET,使用称为EKAREV的CFP-YFP生物传感器(图2)。稳定表达EKAREV的细胞由Somponnat Sampattavanich博士友情提供(图3)。在该生物传感器中,供体和受体荧光团以单一融合蛋白编码。EKAREV生物传感器经过优化,可以减少随机触发的基础FRET信号,并使其可靠地与距离相关。ERK对EKAREV的磷酸化触发构象变化,使CFP和YFP靠近诱导FRET。图2.细胞外信号调节激酶活性报告基因(EKAREV)的示意图。在该生物传感器中,两种荧光蛋白通过ERK底物结构域,接头和结合结构域分开。一旦ERK底物结构域经过ERK的磷酸化,就会触发构象变化,使CFP和YFP紧密接近并允许FRET发生。EKAREV生物传感器是分子内FRET的实例,其中供体和受体以1:1的固定化学计量存在。因此,进行双通道比率实验就足够了,通道1检测受体发射光(IAcceptor),通道2检测供体发射(IDonor),将得到的两个荧光信号强度进行背景校正,并计算它们的比率以给出相对FRET效率EFRET:测定方法将1.2×104EKAREV细胞/孔接种到CellCarrier-96Ultra微量培养板(PerkinElmer#6055300),150μl培养基(表1)中。孵育2天后(37℃,5%CO2),150μl饥饿培养基洗涤两次并在饥饿培养基中孵育5小时以降低基础ERK活性。另外,在孵育开始时向细胞中加入各种浓度的抑制剂或DMSO。4.5小时后,将细胞核用4μM DRAQ5在37℃,5%CO2下染色30分钟。然后用饥饿培养基洗涤细胞一次,并加入含有8μl 20x浓缩抑制剂或DMSO对照的150μl新鲜饥饿培养基。作为对照,在某一时间点,向细胞中加入8μl20x浓缩诱导物(PMA或EGF)。为了抑制FRET信号,应用PD184352,SCH772984和Ulixertinib。含有或不含有所测试化合物的最高DMSO浓度的培养基用作对照。试剂,化合物和介质列表成像在宽场模式下使用20x高NA物镜(NA 0.8)在Operetta CLS系统上建立长时间实验,获取图像总共97分钟。将FRET诱导化合物添加到血清饥饿细胞后,开始时间序列,测量间隔为每8分钟一次,在此设置中获得了四个渠道:DRAQ5 (ex 615-645,em655-760),CFP(ex 435-460,em 470-515),YFP(ex490-515,em 525-580)和FRET(ex 435-460,em 515-580)(图3)。图3.稳定表达EKAREV生物传感器的人乳腺上皮细胞。细胞核用DRAQ5染色。随后,在Operetta CLS系统上使用宽场模式的20x高NA物镜对细胞成像。分析策略使用Harmony高内涵成像和分析软件进行自动图像分析。简言之,将图像分割成细胞和背景。计算细胞质和背景中的供体和FRET强度,然后计算背景校正的FRET比率作为最终结果(图4)。图4.使用Harmony软件进行比率FRET定量的图像分析工作流程:细胞和背景的细胞质被分段,低表达细胞被强度阈值排除。量化供体和FRET通道的强度及其适当的背景,并计算背景校正的FRET强度比。减去背景强度在活细胞应用中尤其有利,其中具有自发荧光组分的培养基通常导致更高的背景并因此导致更小的测定窗口。结果为了探索是否可以使用基于FRET的生物传感器在Operetta CLS上研究ERK信号传导的调节,用不同的ERK和MEK激活剂和抑制剂处理EKAREV细胞。(图5)。图5.外源添加的活化剂(绿色)和抑制剂(红色)示意图及其对ERK信号通路的影响。表达EKAREV的细胞用EGF或PMA处理以诱导ERK活化,另外,用三种MEK和ERK特异性抑制剂(PD184352,SCH772984,Ulixertinib),在途径的不同位置中断信号转导。PMA和EGF充当Ras/Raf/MEK/ERK信号级联的特异性激活剂。EGF特异性结合细胞表面上的EGF受体,而PMA作为亲脂性,膜可渗透的分子通过直接激活RAF激活该途径。PD184352可以通过选择性抑制MEK1/2来抑制ERK途径,而Ulixertinib和SCH772984都是ERK1/2的有效和选择性抑制剂。首先,为了更多地了解FRET诱导和抑制的动态性质,记录了97分钟的长时实验。正如所料,与未处理的对照相比,单独用EGF或PMA处理细胞导致FRET比率的强烈增加(图6)。大约30分钟后信号处于高位。对照显示较低水平的ERK活化,并且观察到随时间稳定增加。由于ERK1/2可以通过多种生长因子和有丝分裂来调节,这可能是由活细胞成像过程中的自分泌或旁分泌信号引起的。用不同浓度的ERK抑制剂(SCH772984)共同处理细胞导致ERK反应的剂量依赖性降低。在5μMSCH772984中,通过EGF的ERK活化几乎可以忽略不计,表明在该浓度下ERK被完全抑制。请注意,0.5%DMSO是实验中使用的最高浓度,确实对FRET比率有影响,因此需要包括此对照。用第二种ERK1/2特异性抑制剂Ulixertinib获得了类似的结果(数据未显示)。图6.在Operetta CLS系统上使用基于EKAREV FRET的生物传感器的ERK信号传导的时间进程。通过EGF或PMA刺激ERK诱导快速FRET信号增加,在约30分钟后平稳。高浓度的SCH772984(5μM)导致几乎完全抑制ERK活化(1μg/ ml EGF),没有可测量的FRET信号增加。较高稀释度的SCH772984仅部分抑制EGF诱导的ERK活化。control显示没有任何处理的样品有中间轻微上升的FRET信号。0.5%DMSO略微抑制FRET信号,这是实验中使用的DMSO的最高浓度。测定统计:Z' = 0.87(在时间点32分钟计算,DMSO为阴性,EGF为阳性对照)当FRET信号在32分钟后达到恒定水平时,选择该时间点以确定SCH772984的IC50值。用1μg/ mL EGF和系列稀释的SCH772984处理EKAREV细胞,稀释范围为10pM至3μM。计算的IC50值为272nM的剂量反应曲线如图7所示。图7.ERK抑制剂SCH772984导致基于FRET的EKAREV信号的剂量依赖性降低。在1μg/ ml EGF存在下,用递增浓度的SCH772984处理EKAREV细胞。在孵育32分钟后,在Operetta CLS系统上测定FRET比率,因为信号在此时间点稳定。高Z' 值(Z' = 0.89)显示出优异的分析性能。为了研究EKAREV FRET成像测定是否可用于研究直接作用于MEK1/2的途径调节,测试了MEK1/2抑制剂PD184352对PMA化细胞的作用(图8)。如图所示,PD184352抑制PMA诱导的ERK活化。图8.在Operetta CLS系统上测量的PD184352对PMA活化的Ras/Raf/MEK/ERK信号级联的抑制。EKAREV细胞用另一组活化剂和抑制剂(PMA+PD184352)处理,其作用在RAF/MEK的上游(与图5比较)。用200或2000nM PMA处理的EKAREV细胞显示出高FRET反应(诱导后32分钟)。通过将细胞与MEK1/2特异性抑制剂PD184352以10μM的浓度共孵育来抑制活化。结论EKAREV FRET生物传感器可用于Operetta CLS系统的活细胞成像测定,以研究ERK的激活和抑制。级联内不同靶标的调节很容易测量,因此这种方法可以有助于鉴定干扰Ras/Raf/MEK/ERK信号级联的新化合物。该测定在活细胞中进行,因此它可用于分析ERK信号传导动力学,而定量ERK磷酸化的常规生物化学技术通常是终点测定。尽管细胞群中生物传感器表达水平相对不均匀(图3),但FRET比率的计算提供了特别好的化验数据和统计数据,Z' 值高于0.87。EKAREV生物传感器的优化设计,Operetta CLS系统的高质量成像以及Harmony内图像分析的出色工具都有助于提高这里提供的高含量FRET分析的稳定性。Harmony软件的构建模块概念允许创建易于设置和理解的图像分析序列,并且不需要专业的图像分析知识。该测定还提供了Opera Phenix™ 高含量筛选系统的可比较结果和测定统计数据。由于Operetta CLS和Opera Phenix系统比传统显微镜具有更高的通量,基于FRET的生物传感器的高含量成像为药物发现和细胞信号传导中的基础研究开辟了新的可能性。参考文献1. Pearson, G., Robinson, F., Beers Gibson, T., Xu, B-E.,Karandikar, M., Berman, K. & Cobb, M. H. (2001).Mitogen-Activated Protein (MAP) Kinase Pathways: Regulation and Physiological Functions. Endocrine Reviews, 22(2), 153-183. doi/10.1210/edrv.22.2.04282. Alberts, B., Johnson, A., Lewis, J., Morgan, D., Raff, M.,Roberts, K. & Walter, P. (2007) Molecular Biology of the Cell,Garland Science., 5th revised edition, ISBN-10: 08153410593. McCubrey, J. A, Steelman, L. S., Chappell, W. H., Abrams,S. L., Wong, E. W. T., Chang, F., Lehmann, B., Terrian, D.M., Milella, M., Tafuri, A., Stivala, F., Libra, M., Basecke, J.,Evangelisti, C., Martelli, A. M., and Franklin, R. A. (2007):Roles of the Raf/ MEK/ERK pathway in cell growth, malignant transformation and drug resistance. Biochimica et Biophysica Acta, 1773,1263–84. doi:10.1016/j.bbamcr.2006.10.0014. F?rster, T. (1948). Zwischenmolekulare Energiewanderung und Fluoreszenz. Annalen der Physik 437 (1-2), 55-75.5. Sun, Y., Wallrabe, H., Seo, S.-A., & Periasamy, A. (2012). FRET microscopy in 2010: The legacy of Theodor F?rster on the 100th anniversary of his birth. Chemphyschem., 12(3), 462–474.doi:10.1002/cphc.201000664. FRET6. Fassler, M., Boettcher, K., Malle, M. (2015): Measuring FRET using the Opera Phenix High Content Screening System: A High Throughput Assay to Study Protein-Protein Interactions,Application Note published by PerkinElmer, In., Waltham,MA, USA7. Komatsu, N., Aoki, K., Yamada, M., Yukinaga, H., Fujita,Y., Kamioka, Y., & Matsuda, M. (2011). Development of an optimized backbone of FRET biosensors for kinases and GTPases.Mol Biol Cell, 22, 4647-56. doi/10.1091/mbc.E11-01-00728. Harvey, C. D., Ehrhardt, A. G., Cellurale, C., Zhong, H., Yasuda,R., Davis, R. J., & Svoboda K. (2008). A genetically encoded fluorescent sensor of ERK activity. PNAS, 105(49), 19264-19269. doi_10.1073_pnas.080459点击链接了解更多珀金埃尔默高内涵相关资料http://e86.me/0ZaJW1关于珀金埃尔默:珀金埃尔默致力于为创建更健康的世界而持续创新。我们为诊断、生命科学、食品及应用市场推出独特的解决方案,助力科学家、研究人员和临床医生解决最棘手的科学和医疗难题。凭借深厚的市场了解和技术专长,我们助力客户更早地获得更准确的洞见。在全球,我们拥有12500名专业技术人员,服务于150多个国家,时刻专注于帮助客户打造更健康的家庭,改善人类生活质量。2018年,珀金埃尔默年营收达到约28亿美元,为标准普尔500指数中的一员,纽交所上市代号1-877-PKI-NYSE。了解更多有关珀金埃尔默的信息,请访问www.perkinelmer.com.cn

























 我要推广仪器
我要推广仪器
 下载APP
下载APP