液相色谱清洗液 液相色谱的清洗液,也就是液相泵柱塞清洗液,大家一般采用10%的有机试剂。有人选择10%的甲醇水溶液,说甲醇水溶液接近流动相,粘度也小,更适应系统。有人用10%的异丙醇水溶液,说10%的异丙醇溶液粘度大,清洗能力强,更适合清洗。也有少数人用10%的乙醇水溶液,说乙醇毒性小,便宜,粘度也小,清洗能力也还不错。当然也有人采用纯净水,说纯净水更便宜,更环保。 大家都知道,液相泵柱塞清洗,主要是清洗柱塞往复运动带出的缓冲液(当然有时也要清洗流动相中的酸、碱性溶液),理论上采用纯净水就可以,但纯净水时间长了会有细菌滋生,污染系统。一般大家都会在纯净水中加入一定量的有机试剂,有机试剂一般加5-20%。5%以上的有机溶液具有较强的杀菌功能,20%以下的有机溶液,缓冲液不至于析出。至于加甲醇还是乙醇还是异丙醇、丙酮那就得看实际需要和实际情况了,各有优缺点。 当然这个清洗液远不止这些。如果是正向色谱,流动相是极性较弱的试剂,那样清洗液也尽量选择极性较弱的试剂,比如正己烷、正丁烷等。有时使用树脂填料类的色谱柱,清洗液中最好不要加有机试剂,用纯净水或在纯净水中加一点酸或碱。这个也得看具体要求和具体情况了。 所以液相色谱柱塞清洗液的选择有一些说法。我们常用的是反相色谱,清洗液一般采用10%的甲醇水溶液,10%的乙醇水溶液,10%的异丙醇水溶液,纯净水等。其它色谱法或色谱柱如有特殊需要,那就得特殊处理。这个得看具体要求和实际情况,折优选择。
方法一:1、倒干色谱样品瓶内试液。2、全部浸入95%酒精,超声洗2次后倒干,因为酒精易进入1.5mL的小瓶,且能与大多数有机溶剂互溶以达到。3、倒入清水,超声洗2次。4、倒干瓶内洗液,于110摄氏度烘1~2小时,绝对不能于高温下烘烤。5、冷却,保存。方法二:1、自来水冲洗几遍。2、放入倒有纯水的烧杯中,超声15分钟。3、换水,再超声15分钟。4、泡在倒有无水乙醇的烧杯中。5、最后取出自然风干。方法三:1、先用甲醇(色谱纯)浸泡,并超声清洗20分钟,后将甲醇倒干。2、再将色谱样品瓶内注满水,超声清洗20分钟,后将水倒干。3、后将色谱样品瓶烘干。方法四:1、一般都是先用清水冲洗烘干后再用硫酸重铬酸钾洗液浸泡。2、色谱样品瓶的洗法和做液相等其他是一样的,首先使用医用酒精侵泡4个小时以上,然后超声半个小时,然后倒出医用酒精,使用水超声半个小时,用水冲洗后烘干就可以了。方法五:1、如果费用充足,每次用新的最好。2、如果要重复使用,清洗的方法也很重要,首先用强氧化清洗液(重铬酸钾)浸泡24小时,然后用去离子水在超声波条件下清洗三次,最后用甲醇清洗一次,烘干即可使用。3、瓶垫一定要换成新的,特别是分析农药残留时,一定要换,否则会影响定量结果。
液相色谱分析间苯二磺酸和苯磺酸的条件及定量方法?
关于液相色谱的定量分析定量分析是在定性分析的基础上,需要纯物质作为标准样品。液相色谱的定量是相对的定量方法,即:由已知的标准样品推算出被测样品的量。
液相色谱管路如何清洗
原文来自:JANUARY 2003 LC[url=https://insevent.instrument.com.cn/t/Mp]gc[/url] NORTH AMERICA VOLUME 21 NUMBER 1 19,20,22,24,26http://www.chromatographyonline.com/反相高效液相色谱柱的清洗和再生(二)http://www.instrument.com.cn/bbs/shtml/20071105/1045750/反相高效液相色谱柱的清洗和再生(三)http://www.instrument.com.cn/bbs/shtml/20071105/1045752/[size=4]本文原是2年前让我的研究生左莹暑假特意翻译的,只是后来忘了。一个月前,在整理电脑资料中发现了这篇文献翻译,我重新校对,修整了一些错误,但仍可能会有些翻译不对的地方,或者语句不畅,请大家指正。[/size][color=#00008B]][color=#00FFFF][size=4]反相高效液相色谱柱的清洗和再生[/size][/color][/color] 这个月的“关注色谱柱”着眼于将一根污染的柱子回到或接近初始状态的实用方法。Ron Majors 会讨论硅胶基质或其它类型的反相柱子的清洗步骤。 反相液相色谱是迄今为止高效液相色谱法(HPLC)中使用最广泛的技术[1]。它的广泛使用是因为它能应用于大部分的非极性化合物、许多可电离的及离子化合物的分析。大部分用于反相色谱的固定相是亲水性的,因此分析物是通过其与固定相之间的亲水作用的程度来进行分离的,含有亲水组成的基团也有相似的保留行为。 表一列举了最常见的键合硅胶的固定相(1)。一些比较次等的固定相比如说一些混合固定相(例如:苯基-乙基),末端封尾和不封尾类型的,一些极性固定相同样属于硅胶键合。其它一些各种各样的填料同样也被用于反相液相色谱,包括聚合物,涂布有硅和铝的聚合物,涂有氧化锆的无机-有机杂化物和石墨化的碳。各种不同的固定相都有其自身的优点和缺点。表1 HPLC固定相中的使用比例*固定相 使用比例C1839C826CN**14.5苯基12C43.7亲水作用1.8C21.2C10.8其它0.8聚合物0.5* 来源于文献1** 包括正相色谱,因为正相与反相使用无法确定比例。 反相色谱柱由于可使用多种流动相和添加剂而得到了广泛的使用。其中使用添加剂的一些技术可改变或修饰填料表面。有时,这些添加试剂本身就会污染填料表面或键合相。 由于使用了亲水性的键合固定相,硅胶表层有了其他的化学特征。残余的硅羟基存在于所有的硅胶键合相的表层。图一描述了可能存在的不同类型的硅羟基(2)。由于是弱酸的特性,这些硅羟基会同某些分析物和基体化合物相互作用,尤其是同一些碱性化合物。因为硅羟基的Pka值大约是在4.5左右。电离可能发生在中间的pH值,因此存在其同阳离子产生静电作用的可能性。比较老的A型硅胶可能含有高浓度的金属离子(通常为100ppm,或更高),这些金属离子会在硅胶表面提供更强的酸性,同样还会和金属螯合化合物(3)相互作用。残留的硅羟基会在末端不封尾的键合硅胶和短链键合的固定相上(例如C2和C4固定相)造成较多的麻烦。 色谱柱的使用者必须清楚色谱柱固定相的表面特征和可能存在的分析物—固定相表面的相互作用,所以在使用和发展反相色谱方法时必须考虑可能存在的基体相互作用。比如一些非常疏水的材料如玉米油、特别芳香类材料和蜡可以附着在反相填料表层并改变填料层的特征。含有蛋白质的生物液体会吸附在填料表层,尽管分析者尽了最大的努力来保护高效液相色谱柱以防止外来物质对其的伤害,最终是,某些分析物-基体化合物还是会对固定相产生不利的影响。 当一根色谱柱被污染之后,它的色谱性征可能已经不同于未被污染的色谱柱了。被污染的色谱柱可能会表现出反压力的问题。对于一根被污染的色谱柱则必须通过清洗和再生将它恢复到原来的操作条件。“关注色谱柱”这部分将会讨论一些实用的方法来将色谱柱恢复或接近到初始状态。由于键合的硅胶柱是最常用的,所以我要着重讨论他们。在最后,我将会论述一些其他类型的反相色谱柱的清洗程序。 反相色谱柱中污染物是怎样的形成? 通常,样品基体里总是含有一些对分析者来说不感兴趣的化合物。盐类、脂质、含脂肪的化合物、腐殖酸、疏水性的蛋白质和其他的一些生物化合物就是在使用中能够接触到高效液相色谱柱的可能物质。这些物质可能会比所需分析的物质更强或更弱的保留能力。那些保留能力比较弱的化合物如盐类,将以死体积被洗脱出来。这些不希望发生的干扰可以通过检测器观察到,它表现为一些色谱峰、斑点、基线干扰,甚至是一些倒峰。如果样品基体组成在色谱柱中有强的保留性,且流动相组成本身就不强到足以使其洗脱出来,这些被吸附的、或是被吸收的化合物将会累积,通常是在经过多次进样之后堵塞在色谱柱的柱头。这种特征是在等度条件下观察得到的。中等保留能力的样品混合物可以被缓慢的洗脱出来,其表现为宽峰、基线干扰或基线漂移。 有时,这些吸附的样品组成达到了比较高的浓度,它们便表现为一种新的固定相。被分析物可与这些杂质作用并表现为新的分离机理。它可能会引起保留时间的变化和峰的拖尾。如果色谱柱受到了比较严重的污染,色谱柱的反压力会达到一个无法承受的高度,这将会使泵超压工作,在堵塞的位置引起柱的塌陷并产生中空。表2分析柱的柱体积柱的尺寸(mm*mm)死体积(mL)250 * 4.62.5150 * 4.6 1.5150 * 3.00.64150 * 2.10.2850 * 4.60.5030 * 4.60.3015 * 4.60.15反相高效液相色谱柱的清洗和再生(二)http://www.instrument.com.cn/bbs/shtml/20071105/1045750/反相高效液相色谱柱的清洗和再生(三)http://www.instrument.com.cn/bbs/shtml/20071105/1045752/
[url=https://insevent.instrument.com.cn/t/5p][color=#3333ff]液相色谱[/color][/url]流动相不应含有任何腐蚀性物质,含有缓冲液的流动相不应保留在泵内,尤其是在停泵过夜或更长时间的情况下。如果将含缓冲液的流动相留在泵内,由于蒸发或泄漏,甚至只是由于溶液的静置,就可能析出盐的微细晶体,这些晶体将和上述固体微粒一样损坏密封环和柱塞等。因此,必须泵入纯水将泵充分清洗后,再换成适合于色谱柱保存和有利于泵维护的溶剂(对于反相键合硅胶固定相,可以是甲醇或甲醇-水)。
三 清洗硅胶基质的色谱柱 恢复一根已受污染的高效液相色谱柱的关键在于了解污染物的性质,然后寻找一种合适的溶剂将其去除。如果污染物是由于一些强保留物质的多次进样积聚而产生,一个除去这些污染物的简单的冲洗过程往往会使色谱柱恢复性能。有时,在经过了等度操作之后,用20个柱体积的90%~100%的溶剂B(二元反相体系中较强的溶剂)将会除去这些污染物。(表二列出了各种型号的高效液相色谱柱的柱体积,所以读者可以很轻易地确定色谱柱的冲洗体积。)例如,脂质类的化合物可以使用一些非水性的溶剂来去除,如甲醇、乙睛、四氢呋喃。如果你正在使用含水的缓冲流动相,则千万不能将流动相直接跳到强溶剂中。将流动相猛然间改到高比例有机相的举动会导致高效液相色谱流动体系的缓冲沉淀,这将会造成更加严重的问题,如使筛板堵塞,接口管路堵塞,泵的密封垫失效,刮伤泵的柱塞杆,使进样阀转子失灵。相反,应使用非缓冲的流动相来冲洗色谱柱(就是用水来代替缓冲液)。在经过了5~10个柱体积的非缓冲溶剂冲洗之后才能允许较强的溶剂流经色谱柱。 有时,流动相中的强溶剂组分是不能充分去除色谱柱中的污染物的。必须另外使用一种更强的溶剂或一系列的溶剂才能清洗色谱柱。假如污染物是非生物性的,则使用者可以通过一种或更多的其他有机溶剂来去除不需要的化合物。可以使用许多种的溶剂和溶剂组合方式。访问一些色谱柱生产厂商的网站可以浏览到一些推荐使用的溶剂系统。 一般来说,所有的清洗都会遵循一个相似的模式。清洗过程中的溶剂浓度是增加的,通常最后都是使用一些非极性的溶剂(例如:乙酸乙酯,乃至碳氢化合物),它将会有助于溶解一些脂质类和油类化合物。重要的是要确保这一系列的每个溶剂都能与所使用的下一个溶剂互溶。一个清洗循环的结论是,在回到初始的流动相系统之前,可通过中间的可互溶的溶剂反向走。例如,异丙醇就是这个中间步骤的一个极好的溶剂,因为它能够同有机溶剂互溶,如正己烷和二氯甲烷,而且同样也能够和水相溶剂互溶。因为异丙醇有很大的粘度,所以必须确保清洗时流速不能太高,否则会引起泵的压力过载。同样,如果使用了紫外检测器,则需避免使用在紫外光谱区有吸收的溶剂,因为这样会需要大量的清洗溶剂才能去除所有吸收的溶剂以得到一个稳定的基线。对于典型的键合硅色谱柱和无缓冲液的流动相的一个推荐使用的清洗系统就是:l100%甲醇;l100%乙睛;l75%乙睛——25%异丙醇;l100%异丙醇;l100%二氯甲烷;l100%正己烷; 当使用了二氯甲烷或正己烷作为冲洗溶剂后,由于溶剂的不互溶性,需要先用异丙醇冲洗色谱柱,而后才能使用含水的流动相。冲洗色谱柱的清洗溶剂的体积最小为柱体积的十倍。对于一根250mm×4.6mm的色谱柱,分析者可以使用经典的1~2mL/min的高效液相色谱流量。为了要回到原来的流动相,色谱工作者可以跳过颠倒使用该系列的清洗溶剂。推荐使用异丙醇为中间的清洗溶剂,然后用不含缓冲液的流动相冲洗,最后再用最初使用的流动相进行冲洗。四氢呋喃是另一个使用广泛的溶剂,它可以被用来清洗受污染的色谱柱。如果使用者怀疑色谱柱受到了比较严重的污染,则可以用二甲亚砜或二甲基甲酰胺与水以50:50的比例,以小于0.5mL/min的流速进行清洗。成功的反向液相色谱柱的再生需要花费相当多的时间,使用溶剂进行冲洗可以设置梯度程序来进行通宵操作。* 在清洗过程中产生了是否应该将色谱柱颠倒过来冲洗的问题。因为大部分的强保留的污染物都会留在色谱柱的前端,将色谱柱颠倒过来清洗会减少已被溶解的污染物流出色谱柱的迁移距离。就填料层的稳定性而言,大部分的现代高效液相色谱柱都是用比普通操作压要高得多的压力装填的;因此,色谱柱的填料层应该不会受到反向的流速的干扰。然而,如果色谱柱顶端的筛板的空隙要比底部的来得大,这种反向的方式则是有害的。比如说,如果底部筛板的空隙为2µ m,则足够容纳装填有平均填料粒径为5µ m的色谱柱。(含有粒径5±2µ m的尺寸分布)。然而生产商往往在色谱柱的顶端安装空隙度比较大的筛板,以防止其被样品或流动相颗粒所堵塞。如果这种筛板的空隙度要比粒径大小分布的最小微粒大,部分填料会经筛板而流出色谱柱,这样就会产生中空。如果色谱柱上有一个箭头来提醒色谱柱的流向,我觉得应该在反向使用色谱柱之前参考说明使用书,浏览生产商的网站,或与技术支持组进行商讨,以确定这是否是一个安全的举动。无论你是否将色谱柱反向使用,最好要将色谱柱同高效液相色谱检测器断开,使污染物或者颗粒留在筛板上而不流经检测池,因为这些物质会污染检测池。 清洗污染的反相色谱柱的频率依懒于有多少不明物质被注射到柱子中,因为反相色谱柱有时在分辨率损失和外来物质的洗脱前可以忍受大量的污染物, 使用者往往等到他们观察到一些异常现象才对柱子进行清洗。然而,长时间累积的污染物会使色谱柱的清洗工作变得更难。正因为如此,如果你知道自己的色谱柱很容易受到脏的样品基体的污染时,我建议定期清洗你的色谱柱。清洗的次数越多,清洗条件也就越简单。反相硅胶基质色谱柱中残留蛋白质的清洗 如果一些如血浆、血清的生物物质留在了反相液相色谱柱上,色谱工作者必须使用一些不同的清洗程序。在大部分情况下,一些比较纯的有机试剂如乙睛或甲醇是不能溶解肽和蛋白质的,所以它们不能有效清洗反相液相色谱柱。然而加入了缓冲液、酸或者一些离子对试剂的混合有机溶剂能够有效清洗这些物质。起初,可以尝试含比较高浓度的溶剂B的流动相来冲洗色谱柱。Freiser和他的同事(4)发现来回反复地梯度洗脱,使用三氟乙酸的水溶液和三氟乙酸—正丁醇可以使污染的反相液相色谱柱再生。Bhadway和Day(5)建议进样100µ L的三氟乙醇到250mm×4.6mm色谱柱可以达到清洗的目的。如果这些方案都失败了的话,推荐使用Cunico和他的同事们(6)的强洗脱液和溶解性的试剂(见表三)。然而,在用这些试剂冲洗色谱柱之前,应该参考色谱柱的手册,或和生产商进行商议,以确保其不会破坏色谱柱的填料。硅键合色谱柱的填料往往能够与这些试剂共存,而聚合物色谱柱可能会因为与特定溶剂结合而使填料产生膨胀或收缩,从而影响到色谱柱的性能。表三 用于HPLC反相色谱柱蛋白质物质除去的清洗溶剂溶剂组成乙酸1%的水溶液三氟乙酸1%的水溶液0.1%三氟乙酸-异丙醇40:60(V:V)(粘稠的,通常降低流速)TEA-异丙醇40:60(V:V)(在三乙胺混合前用0.25N的磷酸调节pH到2.5)尿素或胍的水溶液5-8M(调节pH到6-8)NaCl,Na3PO4,Na2SO4水溶液0.5-1.0M (Na3PO4 pH 7.0)DMSO-水 或 DMF-水50:50(V:V)来自参考文献6。如果使用了早期的一系列溶剂,则必须确保表三中的溶剂与这个系列中的溶剂都是互溶的。异丙醇是一个良好的中间冲洗溶剂。在体系中的清洗体积最少为20个柱体积。由于一些溶剂清洗系统具有一定的粘滞性,所以必须调整冲洗流速以避免产生超压。在清洗完一根含有胍和尿素的色谱柱后,需要用至少40~50柱体积的色谱级的水进行冲洗。对于反相高效液相色谱柱来说使用一些如十二烷基磺酸钠(SDS)和Triton的清洗剂来清洗是不妥的,因为这些化合物会强烈地吸附在硅胶基质的表面而难以去除。这些试剂会影响填料表层,改变填料的性质。然而,分离小组的研究发现,肽合成过程中保护基团和净化剂产物对柱子的污染,可以通过在流动相中注射500µ L的1%SDS溶液以1mL/min的流速进行冲洗(7)。如果接下来使用含0.1%(V/V)三氟乙酸的5%~95%乙睛的梯度,在开始的条件下进行平衡,多肽的分离效果则可恢复。
[color=#00008B][B]该帖为楼主自己整理,帖中所有楼主撰写的内容未经授权不得转载,否则属侵权违法行为![/B][/color]看到很多版友发帖求助讨论准确性的问题,我特意整理了一个关于定量分析误差问题的帖子,希望对大家有所帮助。高效液相色谱定量分析过程可分为样品的前处理、标准品的配制、进样、色谱分离、检测及数据处理等七个步骤。[B]一 误差的主要来源[/B]随着现在市场销售仪器自动化程度的提高,进样、色谱分离、检测及数据处理等实验环节对实验结果产生的误差越来越小,尤其在高效液相色谱定量分析中,实验结果的误差可能主要来源于样品的前处理及标准品的配制。[B]1、样品的前处理 [/B]样品的萃取率是样品前处理时存在的主要问题。当固-液萃取(含柱分离的前处理方法),液-液萃取时,存在萃取率不高且不稳定的问题。尤其在除去蛋白质时,存在变性蛋白质会吸附一些被测组分,导致萃取率降低的情况。通常,萃取率是通过在式样中添加被测成分在萃取的方法评价的。也就是说,被测成分的增加量和液相色谱中的峰面增大成比例关系,通过这种方法可以确定溶液中被测成分的变化趋势。 如果存在萃取率不稳定的问题,就有必要改变萃取的方法,要预先添加内标,然后萃取。这种情况采用的内标,必须与被测物质的化学结构类似,萃取的萃取率才可能相近。如果回收率不但接近100%而且较为稳定,可以证明这种前处理方法较为可靠。在分析中如果能充分考虑以上误差产生的各种原因,才有可能得到精确的分析结果。 [B]2、标准品的配制[/B]本帖中讨论的问题带有普遍性,影响高效液相色谱分析结果准确性的因素较多,仅在标准溶液的配置过程中,可以分为标准物质的称量,溶液的配制和溶液的储存三个环节。 作为标准溶液使用的标准物质其纯度要求很高,应避免使用纯度不符合要求的试剂,作为标准溶液使用的标准物质具有不可替代性,现在市场有销售的HPLC专用的溶剂及各种标准试剂可供实验选择;其次选择与样品浓度要求相适应的天平,应该尽可能使用高精度的天平,这样才能把由于天平使用带来的称量操作误差降至最低。[B]二 消除误差的方法[/B] 要提高分析结果的准确度,必须考虑在分析过程中可能产生的各种误差,采取有效措施,将这些误差减到最小。[B]1、选择合适的分析方法[/B] 各种分析方法的准确度是不同的。化学分析法对高含量组分的测定能获得准确和较满意的结果,相对误差一般在千分之几。而对低含量组分的测定,化学分析法就达不到这个要求。仪器分析法虽然误差较大,但是由于灵敏度高,可以测出低含量组分。在选择分析方法时,一定要根据组分含量及对准确度的要求,在可能条件下选最佳分析方法。[B]2、增加平行测定的次数[/B] 如前所述增加测定次数可以减少随机误差。在一般分析工作中,测定次数为2—4次。如果没有意外误差发生,基本上可以得到比较准确的分析结果。[B]3、消除测定中的系统误差[/B] 消除测定中系统误差可采取以下措施:其一是做空白实验,即在不加试样的情况下,按试样分析规程在同样操作条件下进行的分析。所得结果的数值称为空白值。然后从试样结果中扣除空白值就得到比较可靠的分析结果。其二是注意仪器校正,具有准确体积的和质量的仪器,如滴定管、移液管、容量瓶和分析天平,都应进行校正,以消除仪器不准所引起的系统误差。因为这些测量数据都是参加分析结果计算的。其三是作对照试验,对照试验就是用同样的分析方法在同样的条件下,用标样代替试样进行的平行测定。将对照试验的测定结果与标样的已知含量相比,其比值称为校正系数。 校正系数=标准试样组分的标准含量/标准试样测定的含量 被测试样的组分含量=测得含量×校正系数 综上所述,在分析过程中检查有无系统误差存在,作对照试验是最有效的办法。通过对照试验可以校正测试结果,消除系统误差。[B]4、样品定量分析过程中的误差[/B]样品处理要尽量减少操作者的技术问题带来的误差,样品的稀释次数、稀释工具都是误差的祸根,应尽量减少稀释次数,稀释工具用高准确度的。样品中的干扰组分会直接影响分析的准确度,而且有些组分会损坏柱子。纯化样品的过程尽量少用蒸发至干的步骤(在色谱分析中这一步又是不可少的),正确操作固相柱萃取、纯化小柱使用的步骤,注意提高每一步的回收率,使用内标法也是一个能准确定量的方法。 在手动进样中进样体积至少是样品定量环管体积的3倍,色谱分离程序要使色谱峰的分离度大于1.5,控制流动相、流量、温度等的平稳。流动相的污染都会抬高基线或减少信噪比,分辨率下降,试验条件的变化,如柱退化、不好的流动相等都能引起保留时间变化,引起一个峰或更多的峰不能被鉴别。正确设定仪器参数,选用合理的数据处理参数,用峰面积计算结果比峰高更精确。[B]三 结论 [/B]以上的分析的是我们能尽量控制的误差,还有一些不是操作者所能控制的误差,如被测定组分易分解、组分的含量高低、介质效应等。我们把能控制的误差减小到最低,那你的结果准确度将更高。
液相色谱自动进样器如何清洗?因为我的自动进样器用的磷酸盐缓冲液,比较容易析晶,刚接触到自动进样,想请问液相自动进样器如何清洗?
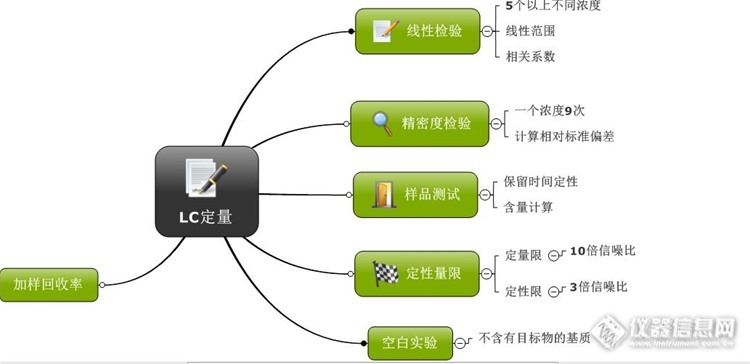
今天读到傅若农教授的文字,感到很温暖。想想我这个分析化学战线的逃兵,将彻底地和分析化学说再见了。但是,我还是讲我的一些思路做一个总结,希望能给我们90后的分析工作者有点启发。接着去年我那个帖子。《谈谈液相色谱方法开发》,有网友希望我写点定量方面的内容。那么,今天,我权当抛砖引玉。当然,我写的很不成熟,希望大家批评指正。http://bbs.instrument.com.cn/topic/5957822在做好了分离方法之后,我们需要对我们的目标物进行定量分析。定量分析的思路一般是http://ng1.17img.cn/bbsfiles/images/2016/06/201606241414_597987_1626663_3.png一般来说,通过这套思路来进行定量方法学的验证。这个是一个通用的方法。当然,这也是十五多年前我做课题时候。 我导师教我的,现在也许也有发展了吧。
[size=4]清洗键合硅胶反相色谱柱的特殊方法[/size]有时,使用有机溶剂是不能去除色谱柱上的污染物的。如果金属离子被硅胶吸附或与键合,这种情况就特别明显了。这时,可以使用螯合试剂如0.05M的乙二胺四乙酸(EDTA)来冲洗色谱柱。EDTA会同许多金属形成络合物,并把他们进行溶解。在使用过了EDTA后,分析者可以用水彻底冲洗色谱柱。如果样品基体含有一些离子性的化合物,可以改变pH值使其变成非离子状态,然后用水—有机溶剂混合溶剂进行冲洗。例如,一个强碱性的基体化合物可以将其pH值调到小于3而将其去除,这会使质子化的胺在水中溶解性更大。对于去除酸性的基体化合物则是将pH值调到比较高—略高于其pKa——pH值大约为8或9,在这个pH值条件下,酸正处于它们的离子状态。然而,要小心键合硅胶基质的色谱柱,因为长时间暴露在高pH值下会损坏的(8)。为了控制缓冲体系和色谱柱中残留缓冲液中的细菌生长,色谱工作者可以使用一些家用的漂白剂稀释到1:10或1:20,用50个柱体积过柱,接着再用50个柱体积的高效液相色谱级的水进行冲洗。不能让漂白剂流经检测器,因为它会破坏流通池。为了防止溶剂瓶中的细菌生长,应该当天配制足够使用的缓冲液,并将不用的缓冲液存放在冰箱中,并加入0.1%的叠氮化钠,不允许在没有流速的情况下使缓冲液长时间驻留在色谱柱中。色谱工作者往往会讨论使用离子对试剂之后对色谱固定相柱产生的影响。一些离子对试剂如辛磺酸(用于阳离子)和四丁基溴化铵(用于阴离子)在含有某些有机改性剂的条件下会强烈地吸附在键合硅相的表面。色谱柱受到了污染不可能再生到他们的初始状态,这只能说用于离子对色谱的色谱柱需要为这门技术而献身,而且永远无法再用于常规的反相高效液相色谱。Bidlingmeyer(9)则并不赞同这种一般性的观点,认为较为极端的pH值,如在酸性条件下(pH 1-3)键合相和末端封尾硅羟基的水解或在高pH值(pH 7-8)条件下的硅胶溶解,这些离子对试剂能改变一些色谱柱的属性。为了去除磺酸类的离子对试剂,他推荐首先使用至少20倍柱体积的不含离子对试剂的相同流动相冲洗色谱柱,然后用不含缓冲液的流动相进行冲洗(在这个清洗步骤中,甲醇是一个比乙睛要好的有机溶剂;对于长链的离子对试剂,需要使用四氢呋喃)很明显,磺酸类的离子对试剂和胺类的离子对试剂存在不同的色谱行为,其对于色谱柱的影响是不同的。Bidlingmeyer和他的同事们(10)证明了当使用了C18柱,流动相浓度大于70%甲醇,SDS,这个长链的阴离子离子对试剂是不会吸附在固定相上的。这种发现也恰好验证了分离组的研究(7)。硅胶键合整体柱,例如Chromolith 色谱柱(Merk KGaA Darmstadt,Germany)应该被当成其他类型的硅胶色谱柱来进行处理。聚合柱的再生用来分离生物分子的聚合柱也会被污染而需要清洗。这种聚合材料的化学稳定性往往决定于它们的强度。实际上,许多的生产商推荐用1.0M的硝酸或1.0M的氢氧化钠来清洗聚合柱。某些反相聚合物柱子如聚乙烯填料(苯乙烯——二乙烯基苯)(PS-DVB)和聚合整体柱如CIM RP-SDVB的叠片柱(BIA Seperations, Ljublijana, Slovenia)以及Swift色谱柱(Isco, Lincoln, Nebraska)可以承受较宽的pH值范围(通常为1~13,有时为0~14),但是,使用者在用一些要求苛刻的有机试剂清洗色谱柱时应该谨慎。根据它们键交联度,当色谱柱暴露在有些有机溶剂中时,会使色谱柱填料产生收缩或膨胀。8—10%以上的高交联度的聚合填料通常具有非常良好的机械稳定性,在水相溶剂中有最小程度的收缩,而在有机溶剂中有最小程度的膨胀。在用一系列溶剂清洗聚合柱之前,最好能够参阅色谱柱手册,或同色谱柱生产商的技术支持部门进行商讨。按照BIA分离要求(11),使用者再生PS-DVB的聚合物整体柱可以通过:l 用10个柱体积的含0.1%的三氟乙酸的异丙醇,以一半的工作流速进行冲洗柱子;l 用至少5个柱体积的100%流动相B以一半的工作流速进行冲洗柱子;l 用至少10个柱体积的100%的流动相A以工作流速重新平衡色谱柱。如果要清洗一根有丁基和乙基的甲基丙烯酸填料的整体柱,可以用10倍柱体积的每份含1.0M的氢氧化钠、水、20%的乙醇溶液和缓冲液,反方向冲洗色谱柱(12),并将蛋白质除去。对于大部分的亲水性蛋白质,使用者应在用水清洗之后加入一个用异丙醇(30%V/V)或乙醇(70%V/V)的清洗步骤。对于微生物污染的清除和钝化,一根PS-DVB整体柱可以用0.5~1.0M的氢氧化钠完全清洗。装填的整体柱应该在室温下用氢氧化钠浸泡至少一个小时以上。用来分离复杂蛋白质如不溶性的细胞膜蛋白、结构蛋白和病毒外壳蛋白的传统聚合填料的色谱柱只需粗糙的清洗条件。例如,清洗这些复杂蛋白质(13)也许要在60 °C条件下用到含3M盐酸胍的50%的异丙醇。肽的合成中来自于固定相树脂的碎片,产生了活性碳正离子,这些碳正离子可以用苯甲醚和硫代苯甲醚来提取。这些碳正离子反应会产生大量的芳香分子,这些芳香分子会在肽的纯化中破坏反相色谱柱。这些污染物在C18柱内有很强的保留,不能用100%的乙睛或甲醇除去。为了清洗这些色谱柱,应该颠倒色谱柱的使用方向,然后用3~5个柱体积的100%异丙醇,3~5个柱体积的二氯甲烷,3~5个柱体积的异丙醇,再回到原来的初始溶剂系统进行冲洗(14)。芳香性杂质的洗脱可以用紫外检测器在260nm波长下进行核实。 氧化锆基质的高效液相色谱柱的再生ZirChrom 分离有限公司(Anoka,Minnesota)已经生产了一系列的氧化锆柱。其生产线也包括了一些反相液相色谱柱,如聚丁二烯、聚苯乙烯、石墨碳等形式。氧化锆要比硅胶更抗pH值,所以涂敷了氧化锆的色谱柱有更高的耐受性,能够经受住苛刻的操作环境,如高的pH值和操作温度。然而由于这些特殊的表面特性,分析者必须保持特定的分析条件以使他们的色谱柱成功地接受各种分析工作。碳酸、氟化物、磷酸根离子都会强烈地吸附在氧化锆柱上。为了将它们从色谱柱中清除,需用50倍柱体积的20%乙睛—0.1M氢氧化钠或0.1M的四甲基氢氧化铵,10倍柱体积的水,50倍柱体积的20%乙睛-0.1M硝酸,再用10倍柱体积的水,20倍柱体积的100%的有机溶剂进行冲洗。对于聚丁二烯和聚苯乙烯柱,色谱工作者可以用甲醇、乙睛、异丙醇或四氢呋喃。石墨碳色谱柱则需要在相同的溶剂中至少加入20%的四氢呋喃(15)。总结反相液相色谱柱会由于一些含强保留成分的样品基体的反复进样而造成污染,尤其是一些分子量高的、亲水性比较强的化合物、如蛋白质这样的生物流动性组成和一些会吸附在硅醇基上的强碱性物质。另外,某些流动相添加试剂如离子对试剂和表面活性剂会吸附在填料表面并改变其性质。被污染的色谱柱会产生糟糕的峰形、假的可重复的保留性、高的反压力和基线干扰。通常一些能够破坏污染物与色谱柱键合或硅胶表面反应的有机溶剂和试剂可以用来清洗色谱柱。 色谱工作者需要小心谨慎,因为这些试剂本身有很强的破坏作用,使用时会造成固定相本身的损坏。此外,通过使用样品的前处理过程,尽可能少地接触到污染物的样品,来防止色谱柱受污染是一个很好的步骤。在所有的应用中,本人强烈推荐使用保护柱来防止色谱柱的污染并对分析柱可能造成的伤害。
液相色谱仪使用时间久了必须进行清洗,仪器在使用过程中会出现各种故障,以下是色谱仪的常见故障及清洗。 色谱仪器控制的冲洗要比手动冲洗更好,进样阀切换到进样位置后,要在此位置上保持一段时间,使流动相充分冲洗样品定量管,这样可以保证有较高的精确度,而且不用另外再冲洗样品定量管。 每进样10次,或20次最好冲洗一下注射针导入口,这样可以保证注射针导入管内充满流体。在注射针插入或拔出的过程中,清洗注射针头或稀释污染这一区域的样品的同时也使注射针导入口和样品溢出口充满流体,从而防止空气进入样品管。 当样品定量管经过充分的冲洗后,可以将旋柄转回取样位置(Load),也可以继续保持在进样位置,到下次取样前才切换回取样位置。在切换回取样位置时,将样品进样针或微量样品进样针从进样阀中拔出。 如果已发现有严重的交叉污染现象,请检查是否有下列原因造成: 1.注射针没有充分插入注射针导入口。2.使用的注射针头长度不够(针头的长度至少5cm),注射针末端不能到达定子表面。3.注射针导入口的污染物或针头密封磨损下来的削片,使注射针末端不能接触定子表面。
怎样清洗液相色谱空柱?
怎样清洗液相色谱空柱?
我用自动进样,定量环为100微升的液相色谱分析样品,现进样量要改为10微升,进样能准确吗?
液相色谱老是有一个杂峰清洗不掉
液相色谱柱应该怎样清洗
请专家帮帮我我用的是岛津LC-10AD型液相色谱仪,今天进样时发现进的样品从六通阀的清洗管流了出来,把定量阀等清洗之后,还是会流出来。怎么办呀?请各位指点一下!!!也可以发给我: planltosurf@yahoo.com.cn谢谢了,急盼!!
1 色谱柱跑干了,处理方法:将贮液瓶装入重蒸水,首先按purge键,将柱前气柱排出,若无法排出,可将色谱柱前拆下,用注射器抽吸。待柱前气体排空后,连接色谱柱,用水高低流速交替冲洗色谱柱,直至柱压稳定为止。 2.基线不稳,有静电,处理方法:检查接地是否良好。 3.峰面积变化非常大,处理方法:检查是否进样阀堵塞。 4.基线突然提高,变粗,处理方法:检查样品池能量,若低于正常值,说明检测器污染。 5.柱压变动范围较大,处理方法:多为进气泡。高低流速交替冲洗。 6.柱效或峰形突然变差,处理方法:更换预柱。 7塔板数低 :色谱柱选择不对,二次保留、峰形不好的影响 8色谱柱易变性:色说柱选择不对,二次保留的影响 9色谱柱寿命短:色谱柱选择不对,样品需预处理(PH7) 10保留值变化:色谱柱平衡不够,流动相比例改变 11出现新的干扰峰:初始分离不够或初始样品无代表性选择波长要从以下几个方面考虑: 1. 检测波长要大于溶剂截止波长。如果所选择的波长下溶剂有很强的吸收当然是不合适的。溶剂有强吸收后,基线抬高,减小检测的线性范围而且会加大基线噪声,对溶质的检测灵敏度下降。 2. 对各组分都要有适当的吸收。一般是不可能做到的。所以只要选择一个对大多数组分都有较大吸收的波长就可以了。 3. 外标法定量时,要选择被测组份最大吸收波长。 254nm 对常见的共轭结构和羰基官能团都有吸收。对饱和基团也有弱的吸收。而对于常见的乙腈、甲醇的透过率很高。是在不知道用什么波长时最理想的试验波长。 (一)涡流扩散(Eddy diffusion)流动相碰到较大的固体颗粒,就像流水碰到石头一样产生涡流。如果柱装填得不均匀,有的部分松散或有细沟,则流动相的速度就快;有的部位结块或装直紧密则流就慢,多条流路有快有慢,就使区带变宽。因此,固相载体的颗粒要小而均匀,装柱要松紧均一,这样涡流扩散小,柱效率高。(二)分子扩散(Molecular diffusion)分子扩散就是物质分子由浓度高的区域向浓度低的区域运动,也称纵向分子扩散。要减少分子扩散就要采用小而均匀的固相颗粒装柱。同时在操作时,如果流速太慢,被分离物质停留时间长,则扩散严重。(三)质量转移(Mass transfer)被分离物质要在流动相与固定相中平衡,这样才能形成较窄的区带。在液相色谱中,溶质分子要在两个液相之间进行分配,或在固相上被吸附和解吸附均需要一定的时间。当流速快时,转移速度慢,来不及达到平衡动相就向前移,这各物质的非平衡移动,使区带变宽。四)动相流速当流速太低时,分子扩散严重,特别是在[url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]中尤为突出。如将理论塔板高度对流速作图,理论塔板高度随流速增加而急速下降,当达到最低值时,流速再加大则质量转移起主要作用,理论塔板高度又加大。在高效液相色谱中,流速稍快影响不大,但在凝胶过滤色谱中,因为物质要渗透到凝胶内部,所以质量转移影响大,流速咖大会降低柱效率。五)固定相颗粒大小定相颗粒越小柱效率越高,对流动相流动的阻力越大,需要加大压力才能使它流动。 1、开泵后压力即升高至最高限度,经过对色谱柱前,色谱柱,色谱柱后的分段测试,确定为检测池堵塞。 2、换检测波长后,色谱峰面积比以前做的小很多,怀疑进样环或管路漏夜,经检查,排除,最后确定为,检测波长的显示值和实际值不符。 由气味、景象和声音可以发现的问题你需要运用你所有的感官去发现液相色谱的问题。你最好养成习惯,每天花上几分钟运用你的感官(除了味觉)来“感觉”你的液相色谱是否存在问题,这样可以帮助你迅速找到问题所在。例如:在你看到漏液之前,你可能首先闻到它的气味。大部分的问题是可以通过眼睛看到。 A、溶剂的气味原 因 解决方法 1、漏液 1、见前 2、溅出 2、a、检查废液瓶是否已满 b、找到溅出的部位并清洗干净 B、热气味原 因 解决方法 1、仪器过热 1、a、检查并调节通风设施 b、检查并调节温度设定 c、关掉仪器,查找维修手册 C、读数不正常原 因 解决方法 1、压力不正常 1、见前 2、柱温箱问题 2、a、检查并调节设定 b、参照用户手册 3、检测器灯失效 3、更换灯 D、灯警告原 因 解决方法 1、压力超出极限值 1、a、检查是否阻塞 b、检查并调节极限值的设定 2、其它警示灯 2、见用户手册 E、警告音原 因 解决方法 1、溶剂泄漏/溅出 1、找到并解决 2、其它警告音 2、见用户手册 F、刺耳的短音或长音原 因 解决方法 1、轴承失效 1、见用户手册 2、润滑不够 2、进行恰当的润滑 3、机械故障 3、见用户手册 进样阀可能发生的问题 A、手动进样阀,转动不灵原 因 解决方法 1、转子密封损坏 1、更换或调整转子密封 2、转子太紧 2、调整转子的松紧度 B、手动进样阀,载样困难原 因 解决方法 1、进样阀安装不当 1、重新安装 2、定量环阻塞 2、清洗或更换定量环 3、进样器污染 3、清洗或更换进样器 4、管路阻塞 4、清洗或更换管路 C、自动进样阀,不能转动原 因 解决方法 1、无压力(或电源) 1、提供恰当的压力(电源) 2、转子太紧 2、调整转子的松紧度 3、进样阀安装不当 3、重新安装 D、自动进样阀,其它问题原 因 解决方法 1、阻塞 1、清洗或更换阻塞部件 2、机械故障 2、见随机维修手册 3、控制器故障 3、维修或更换控制器 HPLC柱压过高: 1.拆去保护柱,看柱压是否还高,否则是保护柱的问题,若柱压仍高,再检查; 2.把色谱柱从仪器上取下来,如果压力仍然不降,则是管路堵塞,须清洗,如果压力下降了,将柱子的进出口反过来接再仪器上,用10倍量柱体积的流动相冲洗柱子,如果柱压仍不降,再检查 3.更换柱子入口筛板,若柱压下降,说明溶剂获样品中含有固体颗粒,若柱压还高,可在进样器与保护柱之间接一个在线过滤器 基线不稳,上下波动或漂移 1。流动相有溶解气体;用超声波脱气15-30分钟 2。单向阀堵塞,取下超声去除堵塞物 3。泵密封损坏,造成压力波动,更换泵密封 4。系统存在漏液点,确定漏液位置并维修 5。流通池内有赃物或者杂质,清洗杂物 6。柱后产生气泡,流通池出液口加负压调节器 7。检测器没有设定在最大吸收波长处 8。柱平衡慢,特别是流动相发生变化时。用中等强度的溶剂进行冲洗,更改流动相时,在分析前用10-20倍体积的心流动相对柱子进行冲洗 1.检测信号出现倒峰,检查检测器与主机相连接是否有松动,可以把下重新连接。 2.夏季气温高,水容易生菌,要求HPLC所用重蒸水必须每天都要更新。 3.由甲醇换到流动相平衡时,压力先稍稍下降,然后慢慢回升,可能是混合池混合效果不好,也可能是B泵轻度堵塞。 4.色谱柱长期不用,应用甲醇冲洗2h以上后,宁上两端螺丝保存。 岛津机器易出现的毛病是: 1、压力不稳:单向阀堵塞,可拆下,用甲醇水超声; 或漏液; 管路过滤器损坏; 2、基线噪音变大:未接地线,可在检测器的外壳外接一根铁丝连地; 灯能量不足,可在funk功能中查找灯的使用时间,或拆开机器外壳,观看紫外灯的强度; 检测池污染,拆下柱子,用无体积连接器连接泵和检测器,用异丙醇小流速冲洗半个小时即可; 流动相污染或进气泡。 3、泵头有盐析出:应经常用5%异丙醇清洗柱塞杆,否则易磨损。 4、进样口漏液:可能是标准针头变形或损坏,使得进样器磨损,不好的针头应及时扔掉。
春节期间液相色谱仪清洗流程:(1)purge Injector:injector用甲醇保存。purge injector的溶液来源于流动相,可以在冲洗完ABCD某一管路完成后进行。若前期使用中流动相中含有缓冲盐,请先用20%甲醇清洗,再用纯甲醇purge保存。eg: A相放置20%甲醇,将A管路比例设置为100%,purge Injector 2~3次,每次3~5min,再将A相放置100%甲醇,将A管路比例设置为100%,purge Injector 2~3次,每次3~5min。(2)将A,B,C,D四个管路用甲醇充满保存。流速:1ml/min。每条线运行时间,5min(3)seal wash:用50%甲醇运行后保存,运行5min。(4)needle wash:用80%甲醇清洗一次,运行时间5min
使用安捷伦高效液相色谱仪,进不同样品时进样针用什么溶液清洗 是用流动相洗还是甲醇洗呢?用甲醇的话是百分之多少的甲醇?我要测的是有机酸 做标准曲线时 进不同浓度的标准酸品之间进样针是否清洗 用什么清洗?
我是准备用生理盐水对硝酸甘油溶液稀释后用液相色谱进行分析,流动相甲醇:水,215nm波长,应该如何清洗柱子呢?还有NaCl会不会在此处有峰,谢谢。
液相色谱恒流泵的清洗应以多长时间为宜?
液相色谱如何进行定性和定量分析?
[color=#444444]液相色谱进行简单的峰面积比定量分析时,如果合成的样品的溶剂是四氢呋喃,而标准品的溶剂是甲醇,而且合成的样品产量很少,请问如何将四氢呋喃转换成甲醇?是否可以通过氮气吹干四氢呋喃溶剂;是否还有别的可行的方法?[/color]
[table=100%][tr][td]我做氨甲环酸的液相色谱分析,用药典的方法,最开始做都没什么问题,出峰也正常,后来因为有几个数据不清楚,想重新做一下,同样的方法,仪器,就不出峰了,请教一下大家是什么原因,谢谢。[/td][/tr][/table]
我用自动进样,定量环为200微升的[url=https://insevent.instrument.com.cn/t/5p][color=#3333ff]液相色谱[/color][/url]分析样品,现进样量是5微升,进样能准确吗? 会不会导致对照品连续进样5针,峰面积忽大忽小?
液相色谱定量大家一般用外标方法么,还是什么别的方法?
[color=#444444]最近在用液相色谱仪做水杨酸的定量,水杨酸用甲醇:水=60:40配制,流动相为甲醇:水(0.01M甲酸铵)=60:40,但是不知道什么原因,水杨酸的面积随着时间的推移一直在变小?另外,问下大家是用什么方法做的[/color]







 我要推广仪器
我要推广仪器
 下载APP
下载APP