要采用液相色谱对染料的降解产物进行分离鉴定,问下各位分离条件怎样优化啊,本人刚接触这个,非常迷惑
[table=100%][tr][td]我现在做液相色谱方法优化,用的是WVD检测器,是浓度型检测器吗?我想改流速,不知道对峰面积的响应有影响吗?查阅有关文献说峰面积与流速乘积是一个常数,我想知道这个常数是一个定值吗?[/td][/tr][/table]
跟大家分享一本书《高效液相色谱方法及应用》第二版,感兴趣的版友可以下载附件查阅,也欢迎补充。全书的目录如下:作 者: 于世林出 版 社: 化学工业出版社 本社特价书所属丛书: 色谱技术丛书 册 数: 条 形 码: 9787502569068 ; 978-7-5025-6906-8I S B N : 7502569065 出版时间: 2005-6-1开 本: 小16开 页 数: 333定 价: 39 元第一章 绪论第一节 高效液相色谱法的特点一、与经典液相(柱)色谱法比较二、与气相色谱法比较三、高效液相色谱法的优点四、高效液相色谱方法发展简介第二节 高效液相色谱法的分类一、按溶质在两相分离过程的物理化学原理分类二、按溶质在色谱柱洗脱的动力学过程分类第三节 高效液相色谱法的应用范围和局限性一、应用范围二、方法的局限性参考文献第二章 高效液相色谱仪简介第一节 流动相及储液罐一、储液罐二、流动相脱气第二节 高压输液泵及梯度洗脱装置一、高压输液泵二、输液系统的辅助设备三、梯度洗脱装置第三节 进样装置一、停流进样装置二、六通阀进样装置三、自动进样器第四节 色谱柱一、柱材料及规格二、柱填料三、保护柱四、柱连接方式五、柱温控制第五节 检测器一、检测器的分类和响应特性二、紫外吸收检测器三、折光指数检测器四、电导检测器五、荧光检测器六、蒸发光散射检测器第六节 色谱数据处理装置一、微处理机二、色谱工作站参考文献第三章 液固色谱法和液液色谱法第一节 分离原理一、吸附系数二、分配系数第二节 固定相一、液固色谱固定相二、液液色谱固定相第三节 流动相一、表征溶剂特性的重要参数二、液固和液液色谱的流动相第四节 二元溶剂体系中液固和液液色谱的保留规律一、溶质保留值的基本方程式二、液固色谱的保留值方程式三、液液色谱的保留值方程式参考文献第四章 键合相色谱法第一节 分离原理一、正相键合相色谱法的分离原理二、反相键合相色谱法的分离原理第二节 固定相一、键合固定相的制备及分类二、键合固定相的性质三、使用键合固定相应注意的问题第三节 流动相一、溶剂的选择性分组二、在键合相色谱中选择流动相的一般原则三、改善色谱分离选择性的方法四、多元混合溶剂的多重选择性五、溶质保留值随溶剂极性变化的一般保留规律六、用线性溶剂化自由能关系(LSER)来表征反相液相色谱中溶质的保留值方程式第四节 新型高效液相色谱的固定相和流动相一、新型高效化学键合固定相二、化学键合固定相分类方法简介三、整体色谱柱四、超热水流动相第五节 离子对色谱法一、分离原理二、固定相、流动相和对(反)离子三、影响离子对色谱分离选择性的因素参考文献第五章 梯度洗脱第一节 基本原理一、等度洗脱二、梯度洗脱第二节 影响梯度洗脱的各种因素一、梯度洗脱时间(tG)对分离的影响二、强洗脱溶剂组分B浓度变化范围的影响三、梯度陡度对保留值的影响四、柱温变化对保留值的影响五、梯度洗脱程序曲线形状的影响六、影响梯度洗脱的其他变量第三节 优化梯度洗脱的方法一、建立梯度洗脱方法的一般步骤二、梯度洗脱中的实验条件第四节 梯度洗脱的图示方法一、二元溶剂梯度洗脱二、三元溶剂梯度洗脱三、四元溶剂梯度洗脱四、用极坐标和球面坐标描述梯度洗脱参考文献第六章 体积排阻色谱法第一节 分离原理一、分布系数二、体积排阻色谱法的特点第二节 固定相一、固定相的分类二、凝胶固定相的特性参数三、凝胶色谱柱的制备及谱图特点第三节 流动相一、凝胶渗透色谱的流动相二、凝胶过滤色谱的流动相第四节 凝胶渗透色谱法测定聚合物分子量分布一、聚合物分子量、分子量分布及测定的意义二、凝胶渗透色谱图的解析及数据处理参考文献第七章 高效液相色谱法的基本理论第一节 表征液相色谱柱填充性能的重要参数一、总孔率二、柱压力降三、柱渗透率第二节 高效液相色谱的速率理论一、影响色谱峰形扩展的各种因素二、范第姆特方程式的表达及图示第三节 诺克斯方程式一、描述色谱柱性能的折合参数二、诺克斯方程式第四节 色谱柱操作参数的优化一、三个柱操作参数的表达式二、HPLC中实用柱操作参数的优化三、柱操作参数优化的图示表达方法第五节 “无限直径”效应和柱外效应一、“无限直径”效应二、柱外效应第六节 超高效液相色谱一、超高效液相色谱的理论基础二、实现超高效液相色谱的必要条件三、超高效液相色谱的应用参考文献第八章 高效液相色谱分离条件的优化第一节 高效液相色谱中色谱参数的相关性一、色谱参数的分类二、色谱参数的相关性第二节 色谱分离条件优化标准的选择一、难分离物质对的峰对分离优化标准二、整体色谱图的优化标准第三节 色谱响应函数和色谱优化函数一、Morgan和Deming提出的色谱响应函数二、Watson和Carr提出的色谱响应函数三、Glajch和Kirkland提出的色谱优化函数四、Berridge提出的色谱响应函数第四节 色谱分离条件的优化方法一、单纯形法二、窗图法三、混合液设计实验法四、重叠分离度图法五、等强度洗脱和梯度洗脱的优化图示法第五节 优化HPLC分离的计算机辅助方法一、实验设计系统二、人工智能系统第六节 高效液相色谱专家系统简介一、专家系统的组成二、专家系统的使用方法参考文献第九章 微柱液相色谱法第一节 方法简介一、微型柱的分类二、微柱液相色谱法的优点和缺点第二节 基本理论一、柱外效应二、管壁效应三、稀释效应四、分离阻抗第三节 仪器装置一、输液泵系统二、进样系统三、柱系统四、检测器系统五、连接管和接头第四节 微柱的制备一、评价微柱性能的重要参数二、影响微柱分离效率的相关参数三、微柱的制备方法第五节 微柱液相色谱的新技术一、纳米液相色谱技术二、超高压液相色谱技术参考文献第十章 二维高效液相色谱法第一节 描述分离体系效能的参数一、峰容量二、信息量第二节 二维高效液相色谱的技术功能一、切割功能二、反冲洗脱功能三、痕量组分的富集功能第三节 二维高效液相色谱的流路系统一、多通路切换阀二、二维高效液相色谱的流路系统第四节 二维高效液相色谱在蛋白质组学研究中的应用参考文献第十一章 建立高效液相色谱分析方法的一般步骤和实验技术第一节 样品的性质及柱分离模式的选择一、样品的溶解度二、样品的分子量范围三、样品的分子结构和分析特性第二节 分离操作条件的选择一、容量因子和死时间的测量二、色谱柱操作参数的选择三、样品组分保留值和容量因子的选择四、相邻组分的选择性系数和分离度的选择第三节 高效液相色谱法的实验技术一、溶剂的纯化技术二、色谱柱的装填技术三、色谱柱的平衡、保护与清洗、再生技术四、梯度洗脱技术五、色谱柱前和柱后的衍生化技术六、样品的预处理技术参考文献符号表
液相色谱分析过程中,我们通常以标准和药典为准绳,进行方法的确定和开发,但是在实际的分析过程中,如果按照标准操作,不出峰或者出峰情况不理想,该如何对方法进行优化和完善?有些时候我们会束手无策,希望各位搞方法开发的朋友提供宝贵的参考意见!这里的讨论前提为理想情况,不涉及仪器问题。

液相色谱-串联质谱条件测定水果中的多农药残留方法优化本法建立了水果多种农药残留量的快速、简便、准确的测定方法,通过对样品前处理方法、仪器检测方法的考察优化,建立液相色谱-串联质谱检测方法。其中以日本制定的“肯定列表”中的“一律标准”最为严格,限量为 0.01mg/kg,而我国的残留限量标准还不够完善,很多农药还没有制定限量标准,其方法的测定低限(定量限)能达到 0.01mg/kg的要求。1、液相色谱条件考察:在方法建立过程中对液相色谱条件进行了考察,主要考察了色谱柱、流动相等。在色谱柱选择时,比较了BEH C18、 HSS T3、 Zorbax Eclipse Plus C18等色谱柱,发现HSS T3色谱柱对甲胺磷、乙酰甲胺磷等大极性农药的色谱保留效果较好,所以选择HSS T3色谱柱进行下一步的研究。在流动相考察时,发现在流动相中加入0.1%的甲酸可以改善多菌灵、噻菌灵等农药的分离,而且加入甲酸可以在电喷雾正离子( ESI+)模式电离时提供H+,提高电离效果,所以选择在流动相中加入0.1%的甲酸,比较了在水相和有机相中均加入0.1%的甲酸、仅在水相中加入0.1%的甲酸两种情况,发现色谱分离及质谱电离无显著差别,为简化操作和便于使用,选择仅在水相中加入0.1%的甲酸。流动相的有机相选择时考察了甲醇、甲醇-乙腈( 1+1, V/V)、乙腈三种情况,发现采用乙腈时色谱柱的柱压较低,色谱分离也较好,但毒死蜱、辛硫磷、特丁硫磷、敌敌畏等常用有机磷农药的质谱响应值低、重现性差,而选用甲醇时可以显著提高这些化合物的质谱响应及重现性,综合考虑后选择甲醇为流动相的有机相。由于此次分析的多农药的化学性质差别较大,从高极性到低极性均有分布,所以色谱分离时需要采用梯度洗脱模式,通过实验考察,最终确的液相色谱条件如下:a)色谱柱: HSS T3柱,长100 mm,内径2.1 mm,粒径1.8 μm,或相当者; b) 流动相:甲醇-0.1%甲酸溶液梯度洗脱,参见表 1。 https://ng1.17img.cn/bbsfiles/images/2020/09/202009211516176216_1219_2166779_3.png!w607x264.jpgc) 柱温: 35 ºC
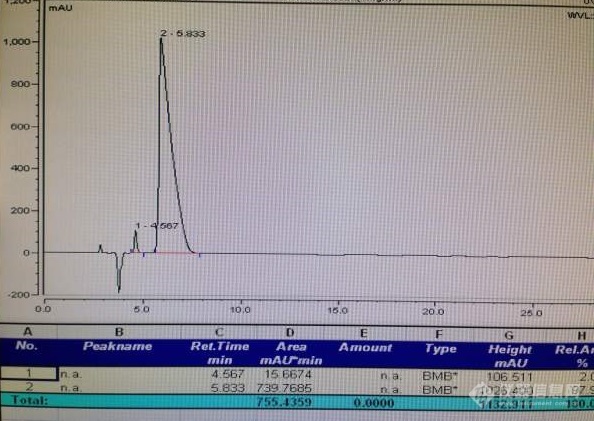
[color=#444444]现想求助一色谱分离问题。[/color][color=#444444] 我做液相色谱时,有两个峰保留时间较近,想让他们分开,大家有什么好方法吗,我已经调过流速、流动相比例和柱温了效果都不明显,具体色谱条件如下,色谱柱:Agilent Eclipse Plus C18(3.5um,4.6*100mm);流动相:0.1%甲酸水:甲醇(95:5),等度;柱温:30℃;流速0.3mL/min;液相色谱图见附件,保留时间为4.567的为图片中的手写结构式,保留时间为5.833的为另一结构式,我想让这两个峰的分离时间达到两分钟以上,因为想让前一个峰进质谱,后一个峰不进,如果浓度降低一起进质谱的话前一个峰就没什么响应了,求高手赐教有什么好办法让这两个峰分的更开一点,谢谢![/color][color=#444444][img=,600,450]https://ng1.17img.cn/bbsfiles/images/2019/09/201909231012429519_6927_1646718_3.jpg!w600x450.jpg[/img][img=,594,421]https://ng1.17img.cn/bbsfiles/images/2019/09/201909231012406386_9342_1646718_3.jpg!w594x421.jpg[/img][/color]
脱氢乙酸具有较强抑制细菌、霉菌及酵母菌发酵的作用,尤其对霉菌的抑制作用最强,是一种高效的防霉、防腐剂。其还具有脂溶性强、热稳定性高的特点,在摄氏120℃的杀菌温度下仍保持杀菌能力不变。国外曾广泛使用于食品、药品中,我国自上世纪70年代中其开始用于食品防腐,曾用于果汁、酱菜、腐乳、干酪、人造奶油、乳酸菌饮料、月饼、果酱等食品。而脱氢乙酸的缺点是毒性较强,目前我国允许在腐乳、酱菜、果蔬汁、肉类制品、糕点、月饼、焙烤馅料中作为防腐剂使用,最大使用量0.5克/公斤。 脱氢乙酸的国标检测方法为[url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]法,样品经过有机溶剂的萃取、净化、浓缩等步骤的复杂处理,并且脱氢乙酸在[url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]条件下,色谱峰出现拖尾现象,使定性、定量影响较大。据报道利用高效液相色谱法食品中的脱氢乙酸,采用纯水、乙醇-水、碱性水对样品进行超声萃取处理,萃取液经过滤后上机检测。作者在试验中发现,利用脱氢乙酸难溶于水而易溶于甲醇、乙醇、乙腈等有机溶剂的特性,样品均浆后酸化处理,用乙腈提取,经微孔过滤后再用高效液相色谱进行定性、定量测定,方法的灵敏度、准确度和回收率高,精密度良好,重现性好,前处理简便快捷,更能满足样品分析要求。 实验样品材料采用一般市售的果汁、酱菜、腐乳、糕点等食品。果汁样品精确称取5.00 g于50 ml的离心管中;酱菜、腐乳、糕点等食品样品事先均匀,准确称取2.0~3.0 g于50ml的离心管中,加入5mL饱和氯化钠溶液和1ml盐酸溶液(1:1),用旋涡混合器混合1分钟,准确加入10mL乙腈,用旋涡混合器混合3分钟,3000转离心15分钟,取上清液经0.45μm过滤器过滤后供液相色谱测定。 按相应的色谱条件对样液进行分析,采用外标法,以保留时间定性,以峰面积定量,测定样液中脱氢乙酸的浓度(mg/ml)。 得到结果以下结果:一、根据扫描的结果,脱氢乙酸的最大吸收波长在230 nm和297 nm处,230 nm处吸收较强,但基体干扰较多,在波长297NM处基体干扰较少,故选取检测波长297NM。 二、动相为乙腈+水时,脱氢乙酸峰形拖尾,使用0.02 mol / L的乙酸铵代替水作流动相,峰形得到较大的改善。乙腈的比例影响出峰的时间和响应,乙腈的比例低,保留时间长,响应也会低,乙腈比例高时,出峰时间短,响应也较高。试验表明,当0.02 mol / L的乙酸铵―乙腈比例为85:15时效果较好。 三、脱氢乙酸难溶于水,易溶于苯、氯仿、乙醚。脱氢乙酸钠则易溶于水,选用水或氢氧化钠溶液、碳酸钠溶液提取,提取液须净化后方可使用。本方法选用乙腈作提取液,主要考虑到脱氢乙酸能溶液于乙腈,乙腈的水溶性有利于乙腈从酸性的样液中把脱氢乙酸完全溶解,同时乙腈可沉淀蛋白质,与脂肪不溶,离心分离得到干净的提取液。 四、在相应的色谱条件下测定,脱氢乙酸的保留时间为5.775min,峰形及组分分离效果好。 五、以70%乙腈水溶液为溶剂配制浓度0.01~0.1范围内的脱氢乙酸标准使用液。以峰面积对脱氢乙酸浓度进行回归分析得回归方程式Y=2.39X×108� 1.33×105,R=0.9997,线性良好。在对同一浓度标样连续进样5次,得到脱氢乙酸的峰面积和保留时间的RSD值分别为0.35%和0.24,方法的定性和定量准确度较高。 本文采用乙腈提取食品中脱氢乙酸,注入高效液相色谱进行测定,以保留时间定性,采用外标法定量。本方法的线性方程有良好的相关性,R=0.9997。方法加标回收率为96.2%~99.6%,变异系数RSD值为1.09%。该方法操作简便、准确,回收率高,精密度良好,重现性好,可用于优化食品中脱氢乙酸的测定。
之前的求助液相色谱检测5种增塑剂用甲醇配的标准溶液出现与dehp重叠的干扰峰怎么解决?重叠峰的问题我找到原因了,应该是之前梯度条件t=0 水:甲醇=30:70t=5 水:甲醇=0:100t=10 水:甲醇=0:100t=15 水:甲醇=30:70太高的问题,这回用0 水:甲醇 15:855 水:甲醇 5:9510 水:甲醇 0:10015 水:甲醇 5:95,20 水:甲醇 15:85 波长225,要是波长275峰就会比225的要低 做校正曲线时就不会出现有重叠峰的错误警告了,但是这个梯度在检测2,5,10.20,50,微克每毫升时峰型还理想,0.5和1就基本没型了,从2到50这五个点做的校正曲线相关系数都在0.99858到0.99976之间,还有我用甲醇配的标准溶液,进样时没有用一次性针头和滤膜过滤,要是过滤的话就会在DEHP的位置出现一个很高很高的峰,就连只进甲醇或者丙酮溶剂都会出现,不知道是不是一次性针头和滤膜中有DEHP?难道就这样过滤下就能污染了吗?要是不过滤又怕损坏柱子,还有这个梯度应该怎么计算啊?也不能就随便尝试没个依据的,看那些理论的线性梯度、凹形梯度、凸形梯度和阶梯形梯度也不知道是怎么算的,有没有简单点的算法啊,或者根据我这次的条件怎样来继续优化,我做的5种增塑剂的保留时间:2.215, 2.791. 4.463. 9.421. 10.024,怎样才能让后两个也早点出峰,不要让他们距离这么远啊,改成下面条件时0 水:甲醇 15:854水:甲醇 5:958水:甲醇 0:10010水:甲醇5:95,15 水:甲醇15:85 保留时间为2.218, 2.783, 4,376, 8.625, 9.164要是把时间在改短些最后一个峰结束后基线会有很大波动,要是运行序列的话,是不是必须要把结束时的梯度设的和初始的一样才能不会影响下一个测试啊?不知道大家都是怎么做梯度优化的?我看了一些论文:如:反相高效液相色谱中复杂体系二元多台阶梯度分离条件快速优化方法 单亦初 赵瑞环 张维冰 梁 振 张玉奎 “提出了一种反相高效液相色谱中二元多台阶梯度分离条件快速优化方法。通过数次线性梯度初始实验,求得溶质的保留方程。在此基础上,利用重叠分离区域图(OSRM) 方法,快速求得复杂样品的最佳多台阶梯度分离条件。该方法只需要几个小时就可以完成对复杂样品分离条件的优化,并通过对中药川芎提取物的分离加以验证,获得了较好的预测精度和分离效果”那些基本原理公式太复杂了,而且他那些梯度具体是怎么计算预测的也没写,希望大家帮帮忙谢谢了
液相色谱分离的峰,每一个峰都只是代表一种化合物吗?还是有可能是几种化合物的混合?峰没有分开是因为流速大还是流速小?流速是怎么影响峰的?谢谢。
液相色谱分离条件的优化可以改变哪些因素
制备型高效液相色谱系统的应用领域制备型高效液相色谱系统主要应用在植化、合成、制药、生物及生化等领域的产品的提取及纯化工作中。在不同的工作领域中,组份的提取和纯化量的差异是很大的。在生物技术领域中,酶的分离是微克级;在植化和合成化学领域中,为了鉴别未知成份并进行结构测定,需要得到一至若干毫克的纯品;在药品和医药学测试中,需要克级的标准品和对照品;在当今的工业级提纯中,制药成份往往需要千克级的提取。制备型高效液相的应用领域可以归纳在下表中。 成份量:所在领域 微克: 生物技术领域的酶的分离、生物学和生化学测试 毫克: 结构描述和特征鉴定,包括:生产中的副产品、生物矩阵的新陈代谢产物、天然产物 克级: 对照品(分析标准)毒物学分析所需组份:高纯品中的主要成份、副产品的分离提取 千克级:工业规模生产,活性成份,药物 制备方法的发展和扩大规模的计算 在分析液相中色谱柱的典型进样量是微克级,甚至更低。样品量和固定相之比有的甚至小于1:100000。进样体积一般来说都大大小于色谱柱体积(小于1:100)。 在这种条件下,会达到很好的分离效果,峰形尖锐并且很对称。而在制备液相中,最大的区别就是超量进样。其结果,超量进样的方法和分析方法的放大将在下章内介绍。 吸附变化线 分析液相的目的是给一种组份定性、定量。重要的色谱参数有溶解度、峰宽和峰的对称性。如果进样量越来越多,峰高和峰面积会增加,但峰的对称性和容量因子保持不变。如下图。 在分析液相中,最佳的峰形应是一条高斯曲线。峰的标准背离 бV 描述了其对称性和与高斯曲线的相似性。容量因子是与一种不保留物质的保留时间t0相关的保留时间。 如果将超过一定量的样品注射进色谱柱,吸附变化线就会成非线性。这意味着峰形会变的不再对称,表现为严重的拖尾和容量因子的缩小。如下图。在制备液相中,这种效果称作浓缩超量进样。在一些情况中,根据进样量的增加,容量因子也相应变大,并造成很强的前峰。既然吸附变化线取决于组份的多少,那么液相色谱柱的载样能力就必须根据不同的制备液相实验来决定。 色谱柱载样和超量载样 大样品量的纯化有两种可行的方法:分析系统的放大或色谱柱超量载样。分析系统的放大意味着使用直径更大的制备柱、更高的流速和根据色谱柱的长度增加进样量并保持样品浓度不变。峰形仍会保持尖锐而对称。这种方法需要大型的色谱柱和大量的溶剂来分离较少的样品,因此这种方法是不经济的。 因此色谱柱超量载样,暨在相同的分析条件下超量进样通常是一种很好的选择。使用色谱柱超量载样的方法,在分析柱上甚至可以进行毫克级的分离。但更大 量的样品分离就需要整个系统的放大。色谱柱超量载样可以通过两钟方法进行— 浓缩法和体积超载法。 在浓缩法中,样品的浓度会提高,但进样体积保持不变。容量因子k’降低,同时峰形从高斯曲线变为矩形。如下图。浓缩法超量载样只有在样品组份在流动相中具有良好的溶解性的条件下才有可能采用。 如果样品组份的溶解性很差,浓缩法超量载样不能使用。同时更多的样品体积注射到色谱柱中,这种技术称作体积法超量载样。超过一定的进样体积,峰高不变,但峰变宽并且呈矩形。在制备液相中浓缩法超量载样比体积法超量载样更受欢迎,因为可被分离的样品量更高。既然组份的溶解性通常是一个限制因素,所以两钟超量载样技术通常被结合起来使用。两种技术的概览浓缩法超量载样 体积法超量载样 取决于组份在流动相中的溶解性 取决于进样体积 吸附变化线的制备部分 吸附变化线的分析部分 生产效率决定于选择性 生产效率决定于制备柱直径 受固定相粒度大小的影响不大 需要小颗粒填料 方法的放大 浓缩法超量载样和体积法超量载样都会导致组份溶解性的降低。既然组份的分离需要一定的溶解性,那么在放大分析方法的时候,优化溶解性、特别是选择性就是一项很重要的工作。 因为选择性和超量载样潜力是相互依靠的,选择性的提高会提高一次运行中所分离的样品量,因此从分析方法到制备方法的放大和方法的优化需要三个步骤。 1. 优化分析方法的选择性。2. 在分析柱上进行超量载样。3. 放大到制备柱 制备型高效液相色谱的目的 判断制备型高效液相色谱使用的结果有三个重要参数:产品的纯度、产量和生产效率。三个参数之间是相对独立的,因此很难同时使用这三个参数来优化制备型高效液相色谱方法。见图形6。 色谱图1显示在制备型高效液相色谱的使用中有很高的生产效率,但是两种组份的分离效果却是很差的。这种方法很可能得到两种组份的高纯品,但是产量和收率却是很低的。 在色谱图2中峰有很好的分离,因此这种方法可以得到两种组份的高纯品和高产量,但是生产效率却很低。 色谱图3中的情况是三个参数综合后得到的最优化的结果。峰在基线上被完全分开,这使得产品纯度、产量和生产效率都达到最高。 在实际应用中,每个参数的重要性都是不同的。如为了进行活性或药物测试,某种组份必须被完全单独提取,那么组份的纯度是最重要的参数,产量和生产效率是其次的。如果某种合成中间体必须被纯化,并且需要有足够的量为下一步合成作准备,那么纯度就不是最重要的了。而生产效率在这种情况下就是个首先需要解决的问题,因为其直接关系到完成整个合成工作的进程和速度。同时产量也是很重要的,因为高价值组份的损失需要控制在最少的范围内。
液相色谱大峰前小峰怎么分离[img]https://ng1.17img.cn/bbsfiles/images/2019/08/201908061642384749_7239_3963683_3.png[/img]
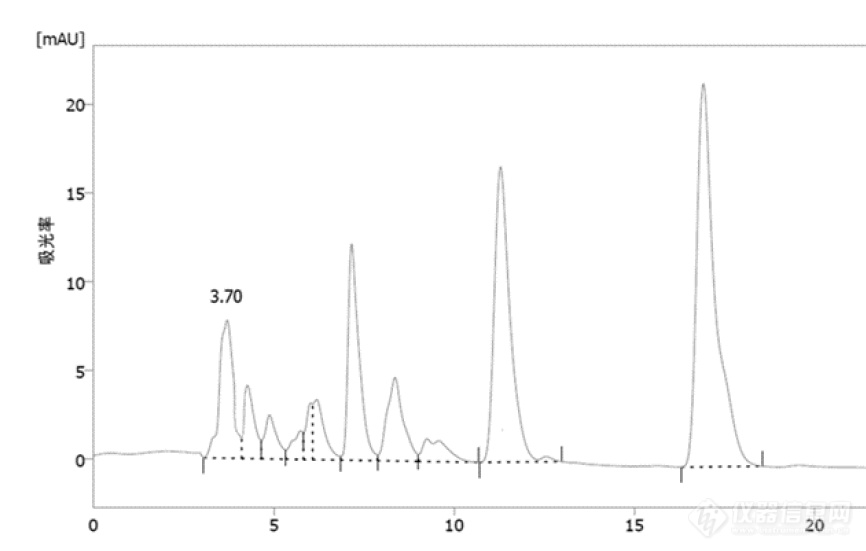
如色谱图,3.7min是我的目标峰(NMN,烟酰胺单核苷酸),怎样优化分析方法来让目标峰更清晰,减少和右边峰的重叠。现在的方法是,A液:pH6.6,50mMNaH2PO4,B液纯甲醇,A:B=95:5,进样量10μL,柱温30℃。[img=,690,439]https://ng1.17img.cn/bbsfiles/images/2021/05/202105101756433478_7214_5263207_3.png!w690x439.jpg[/img]
液相色谱图的优化 我刚刚开始用液相色谱,用的是lc1200 怎样进行报告的设计、数据采集、数据分析、校正表的设计、谱图优化,先谢谢各位前辈了!!!
[b][font=宋体]解答:[/font][/b][font=宋体]([/font]1[font=宋体])化学合成的肽产品是一个纯度不好的粗产品,其一是因为在合成肽过程中各种副反应、消旋化等造成的副反应肽;二是在脱保护过程中,由于保护基的残留,肽键的断裂、烷基化等造成的杂质。由于杂质与合成的肽在分子结构和化学性质上非常相似,给肽的分离纯化带来了困难,需要根据对目的肽的要求选择适当的方法进行纯化。[/font][font=宋体]([/font]2[font=宋体])目前用于分离纯化合成多肽的液相色谱模式主要有三种:一是凝胶过滤色谱,按照肽分子的大小进行分离;二是离子交换色谱,按肽分子所具有的带电基团的性质和数目进行分离;三是反相色谱,按照肽分子的疏水性强弱进行分离。[/font][font=宋体]([/font]3[font=宋体])通常采用制备或者半制备色谱对合成多肽化合物进行分离纯化。制备色谱的进样品量很大,因此要求进样系统能满足大体积进样要求,例如计量泵、定量环的体积会不同于普通分析型色谱。进样量的增加导致制备色谱柱子的分离负荷相应加大,也就必须加大色谱柱填料,增大制备色谱的直径和长度。如[/font]GB/T 20770-2008[font=宋体]《粮谷中[/font]486[font=宋体]种农药及相关化学品残留量的测定[/font][font=宋体]液相色谱[/font][font=宋体]-[/font][font=宋体]串联质谱法》中使用的色谱柱要求柱长[/font]400mm[font=宋体],内径[/font]25mm[font=宋体]。要将待分离组分从如此规格的色谱柱中洗脱下来,就需要使用相对多的流动相,因此制备色谱的管路也比分析色谱粗很大,同时泵的流速也较高,通常需要设置为[/font]5mL/min[font=宋体]。由于制备或者半制备色谱仅用于分离纯化,不用于定量或定性分析,对泵的精密度、检测器的灵敏度等要求较低。[/font][font='微软雅黑','sans-serif'][color=black][back=white]领取更多《实战宝典》请进:[url]http://instrument-vip.mikecrm.com/2bbmrpI[/url][/back][/color][/font][font='微软雅黑','sans-serif'][color=black][back=white] [/back][/color][/font]
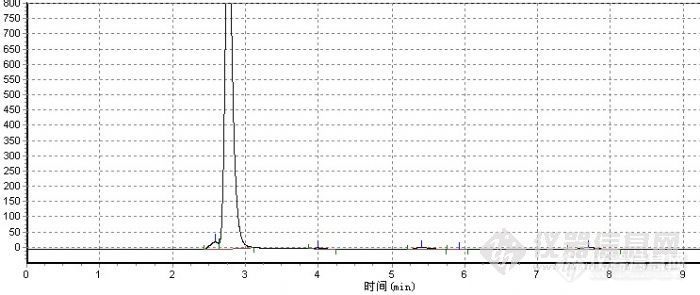
液相色谱检测条件:波长:365;柱温:40℃;流动相:甲醇;流速:0.8;灵敏度0.01; 称重:0.015g;溶剂:100ml三氯甲烷; 进样量:5ul.http://ng1.17img.cn/bbsfiles/images/2010/10/201010151639_251672_2166859_3.jpg各位我现在想请教你们一个问题,我这个主峰是在2.8几出峰的,我现在想他往后推迟,与溶剂峰分开应该怎么做?而且该样品应该有2个峰,该怎么分离出,条件怎么改··谢谢~~!
在测试中心液相色谱组呆了20多年,见过无数的客户,不同的客户对色谱的理解不一。很多人认为色谱就是打一针,出个谱图而已,容易的很,跟他们的科研档次无法比。觉得样品给你了,你必须做出来,而且很快得到其所需要的结果,全然不管你的仪器能否满足要求。做不好或者不想做,就会去告状,服务态度不好之类的。所以一旦征求意见,必然出现各种各样的服务问题。测试人员的地位在学校可见一斑。也有少数本身做液相色谱的,这些人沟通起来非常融洽,因为知道其实际做起来不容易,可惜这类的客户仅有百分之几。在色谱分析的三大分支中,我对液相、离子熟悉,对气相只是很了解,没动手做过。对于这三大类型,其实在实际中做的差别是很大的。先以我精通的离子色谱而言,我几乎涉及了所有的类型,离子色谱能否做关键在于设备,常规的很简单,特殊的全靠设备支撑,因为大部分离子色谱的分析都是优化的方法,固定的模式做起来并不难。但非常规的样品,则难度大大增加,即使同一个组分,由于基体差别,分析方案也是千差万别。也就是常规的很简单,特殊的则很难。现在厂家离子色谱方法开发就是一种特化的过程。离子色谱主要分析离子以及一些极性的化合物,表面上看应用范围比较窄,其实在很多领域有很好的应用,可以解决液相色谱无法解决的一些问题。对于气相色谱,复杂性比离子色谱要高,因为被测的有机化合物种类大大增加,但从气相的结构看,其载气的选择是非常有限的,主要靠色谱柱(极性,非极性,弱极性等),分离则依赖温度的程序升温,它真正的变化在色谱柱,在一般的分析中,大多变化在温度,柱子的变化并不多。因此对于气相色谱,基本就几根柱子。当然一些特别的检测则需要更高级特殊的装置,这跟离子色谱一样。由于受沸点的制约,气相色谱的应用受到很大的限制。而对于液相色谱,就我20年的经历,我认为其复杂性远远高于离子色谱和气相色谱。因为其变化比前二者更多,一是液相色谱分离有很多机理,每种机理都有对应的色谱柱类型,液相色谱的分离机理大约有十来个,很少有人会用过全部机理类型的色谱柱。虽然反相是最常见的分离手段,但由于反相的广泛使用,C18柱的变化类型极多差异很大,这不同C18柱之间的差异有时不亚于不同机理之间的差异。二是,液相色谱最大变化是流动相,不仅有机相类型有变,添加剂类型和浓度有变,不同pH差别很大,面对变化无穷的样品,这个流动相选择变化规律全靠长期的经验积累,很难用文字一言以蔽之。三是,液相色谱的检测器类型最多,离子色谱就三种,气相四五种,而液相色谱的检测器有十来种,相互之间差别极大,不同的检测器对色谱分离机理也有很大的选择性。因此要做好一张液相色谱图,很多情况下只能是你现有条件下的最佳分离,并不是这个化合物的最佳分析条件。对于特殊样品的分析,液相色谱更多的依赖于检测器和柱子的变化,同离子色谱不同。给你一个样品,用那类色谱(液相、气相还是离子),什么柱和条件,则完全依赖你的功底和阅历,当你拥有尽可能多的仪器装备,你才能充分发挥你的能力,依据化合物的特点,样品的特性,选择合适的仪器和配置,做出最佳的色谱图。
分子大小组成相似,分子大小相近,如何用液相色谱法分离分级?液相色谱中哪种方法更好呢?
中药是一个典型的复杂未知样品,其化学物质基础的阐明是中药现代化的关键科学问题之一。液相色谱是中药分析中应用最普遍的技术,也是进一步与质谱、核磁共振等联用的基本分离手段。本论文在总结了中药复杂体系的特点和分析中需要解决的问题后,提出了中药分析的思路,开展了线性梯度下的液相色谱保留值规律、峰形规律的研究,发展了快速获取液相色谱保留参数、峰形参数的方法,从而将未知样品快速转化为色谱已知样品。并以实际样品藏茵陈和黄芪为例,应用发展的方法建立了相应的液相色谱参数数据库,提供中药活指纹图谱的信息基础。同时,面向实际应用,发展了中药组分的目标优化方法。建立了液-质联用表征黄芪皂甙的方法,建立了高效液相制备色谱快速制备黄芪组分的方法并初步发现了具有诱导分化白血病细胞的组分。发展了用五次线性梯度快速、准确地获取液相色谱保留值方程的方法。以线性梯度a、c 值预测任意梯度下的溶质的保留时间,并发展了基于内标峰的校正方法。应用于藏茵陈和黄芪样品的色谱保留参数获取,对c-a 关系进行了考察。基于EMG模型和自动曲线拟合技术,首次建立了以线性梯度快速获取液相色谱液相色谱峰形参数和峰形规律的方法。以线性梯度峰形规律预测了任意梯度条...[img]http://www.instrument.com.cn/bbs/images/affix.gif[/img][url=http://www.instrument.com.cn/bbs/download.asp?ID=31320]中药高效液相色谱分析方法发展的理论基础研究[/url]
今天看了版友一片枫叶的帖子http://bbs.instrument.com.cn/shtml/20141214/5571906/,个人想法与其区别较大,在此我仅说说自己的想法和经验,希望大家多提意见,多交流。建立一个色谱方法应从了解样品特点,确认检测需求入手。了解样品使你清楚方法建立的难点在哪,明确检测需求使你有方法建立的方向性指导,比如建立一个痕量检测的方法紫外检测器通常是不能满足要求的。首先我也讨论一下波长的选择,如果大家在低波长范围内进行测试,那么确实是波长越低样品信号强度越高的,但是在方法建立中样品的检测波长一般以目的物的最大吸收波长为主要参考值,因为很多物质(如实际样品中的杂质)都会在低波长有紫外吸收,采用低波长检测会对真实样品的测试带来不便。选择目的物的最大吸收波长一定程度上可以减少真样品测试时的干扰,而这些干扰在使用标准品进行早期方法开发的时候是不易察觉的。对于流动相的优化一般是方法开发的主体,包括是否使用梯度,pH值,所加盐种类和浓度,所选有机相种类,是否需要添加剂,等。核心目的是所建立的方法对目的物和干扰成分有充分的分离,方法稳定可重复,峰型良好,基本上属于系统适应性考察的内容。降低有机相比例是比较简单的增加样品分离度的方法但会造成样品峰的展宽和整体保留时间的延长,应综合运用各可选条件对方法进行优化,理想的分离时间应使目的物的保留时间在三到五倍的死时间范围内。对于流速的选择我个人认为主要有两个依据:首先是所选色谱柱根据范氏方程估算的最优线性流速,第二是在实际操作中系统的压力。为了保证系统的安全稳定运行可以适当降低流速。但是我并不推荐在压力较低的情况下提高流速而提高分离时间,这样可能造成体系交换不充分而形成色谱峰畸形,过短的分离时间也会使方法的稳定性(尤其是时间稳定性)表现不佳,柱效也会明显降低。作者一片枫叶认为流速增大样品分离度降低,这个现象在样品保留时间比较短的情况下是比较明显的,而在样品保留时间超过三倍死时间后其影响就比较小了,降低流速对样品分离度的影响就比较小了。关于流速对样品检测灵敏度的影响要区分两种类型的检测器,即质量型检测器和浓度型检测器。类似紫外检测器的浓度型检测器其样品峰高与浓度相关,改变流速对峰高理论上无影响,实际工作中流速过快样品峰高会有微弱降低。对于质量型检测器(蒸发光散射检测器可近似看做质量型检测器)降低流速样品峰信号高度会明显下降。对于色谱方法的评鉴应以:柱效、分离度、峰型以及稳定性为核心,对于方法确实对样品的检测灵敏度有影响但检测灵敏度的改善更多依靠于检测器的选择,而检测器对于方法的接受程度是比较高的,特定的检测器有其自身要求,如示差检测器、蒸发光散射检测器、质谱等,在选择这些检测器的时候,其方法建立有特殊要求。关于色谱方法对灵敏度的影响我补充两句:一般同样的样品在不同的溶剂中表现出的响应强度是不同的,另外使用高柱效的短柱代替低柱效的长柱一般会得到更好的样品响应强度
求助液相色谱分离甲酸甲醇乙酸乙醇方法,分不开甲酸和乙醇,现在流动相是磷酸二氢钠和乙腈,C18色谱柱
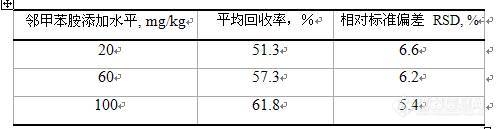
看见同行(看着这话题,绝对是同行,多交流)发着文章http://bbs.instrument.com.cn/shtml/20101012/2857370/,我这把手上的东西整整,算是补充吧。大家多多提意见吧!摘要 针对纺织品偶氮染料检验中常检出的禁用邻甲苯胺及其异构体,结合气相色谱-质谱和高效液相色谱-二极管阵列检测器(HPLC/DAD),摸索了以气相色谱-质谱进行初期定性筛选、以高效液相色谱进行确证和定量的芳香胺异构体检测方法,避免了检验中其异构体造成的假阳性检出。本方法在2 μg/mL~100 μg/mL浓度范围内线性关系良好,线性相关系数可达到0.9999;回收率为51%~62%。关键词 纺织品;检测;禁用邻甲苯胺;异构体1 前言采用气相色谱-质谱联用方法检测染料产品中24种有害芳香胺含量,按照现行国家标准中提供的气相色谱条件,会有多种异构体保留时间相同或非常接近,而且这些异构体的质谱图又非常相似,从而造成无法使用特征离子对其进行定性和定量,往往会形成假阳性结果的产生。当前针对有害芳香胺的气相色谱/质谱检测方法,大都采用非极性或极性较弱的色谱柱,如HP-5MS、DB-5MS、DB-35MS,这些色谱柱普遍存在的缺点是对常见的异构体不能很好的分离。有文献报道采用中等极性色谱柱DB - 17MS(固定相等同于50%苯甲基聚硅氧烷),对芳香胺异构体有一定的分离能力,但没能实现禁用芳香胺的基线分离,造成定量上的偏差。高效液相色谱法(HPLC)可同时测定多种芳香胺,在分离异构体上是强有力的确证方法,准确定性和定量,分析方法易于推广。本方法采用气相色谱-质谱联用对异构体进行初期定性筛选、以高效液相色谱进行确证并外标定量,对常检出的的禁用邻甲苯胺及其异构体检测进行了研究。本方法通过进一步摸索优化高效液相色谱条件,使邻甲苯胺及其异构体达到基线分离,有效避免了检测过程中的假阳性检出。2 实验材料与方法2.1 仪器和设备气相色谱/质谱联用仪:美国Agilent 公司7890A /5975 C,带自动进样器,四极杆质谱检测器;气相色谱柱:HP-5MS 5% Phenyl Methyl Silox,325 °C:30 m x 250 μ[size=
[color=#444444]如题,做液相色谱流动相的优化,有几个问题想请教各位大神![/color][color=#444444]1、主物质峰,最高的那个峰,峰前端有小的凸起,而且峰的前半部分也有凸起的迹象,是不是有两种物质?大神有办法分开不?[/color][color=#444444]2、色谱保留时间普遍偏短,调整流动相比例之后完全没有理想的效果,求大神支招。[/color][color=#444444]3、第一个峰有拖尾迹象,大神可有好的解决办法?[/color][color=#444444][img]http://muchongimg.xmcimg.com/oss2/img/2018/0912/bw133h8159266_1536713421_818.jpg[/img][/color][color=#444444][img]http://muchongimg.xmcimg.com/oss2/img/2018/0912/bw133h8159266_1536713471_269.jpg[/img][/color][color=#444444][img]http://muchongimg.xmcimg.com/oss2/img/2018/0912/bw133h8159266_1536713471_594.jpg[/img][/color]
[align=center][b]浅谈超高效液相色谱柱和液相色谱柱的区别[/b][/align]从色谱柱填料的发展大趋势来讲,色谱柱填料一直是超更小粒径微粒的方向发展的,2004年超高效液相色谱应运而生。更小的粒径,意味着更高的泵压。在色谱柱的发展历程中,泵压、粒度、柱长、柱效这四个因素是相互制约的。随着技术的发展超高效液相色谱可以提供的泵压为140Mpa,而大多数液相色谱仪的最大压强仅为40Mpa。泵技术的突破为超高效液相色谱的产生奠定了基础。两者的区别如下:1、UPLC比HPLC有更好的分离度;我们不难理解填料粒径变小以后,分离度会变好。1.8um颗粒的分离度比5um颗粒提高了70%;2、UPLC比HPLC有更高的分析速度;常规的液相色谱通常在20分钟内完成分离,而超高效液相通常情况下5分钟内就可以完成分析;3、UPLC比HPLC有更高的灵敏度。分析过程中峰展宽变窄,峰高会更高,提升了色谱检测的灵敏度。单纯从色谱柱填料来讲,主要有三个方面的区别 1、Kromasil提供给超高效液相色谱柱的填料粒径为1.8um,普通色谱柱通常填料的粒径为5um。作为液相色谱来说,填料硅胶球的尺寸分布要求非常严格,填料的粒径越小,实际上对硅胶球的要求更高。所以超高效液相色谱柱的填料成本也是高于液相色谱柱的。2、超高效液相的柱压更高,要求填料的抗压性更强,这就要求在填料合成过程中要考虑抗压性的影响,无疑增加了合成难度。3、色谱柱的装填也更加严苛,超高效液相对装填工程师的要求也更高。超高效液相的柱管和柱塞板也是升级和优化过的。
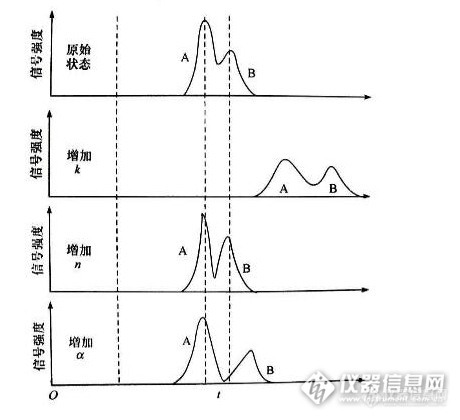
在试样反相高效液相色谱分析中,条件的优化与选择尤其重要,改变不同的仪器操作条件会得到不同的效果,下面是本人在液相色谱分析中得到的一点心得,分享一下,不妥之处请各位版友多多指教!1:试样品分析对反相色谱柱的选择 不同的反相色谱柱,在柱温 波长 流速以及流动相配比 相同的条件下:分离度不同----表现为各组分的保留时间不同。灵敏度不同----表现为峰高和峰面积不同。(下图引用自“中国药科大学色谱分析课程”)http://ng1.17img.cn/bbsfiles/images/2014/12/201412190919_527878_2960432_3.pnghttp://ng1.17img.cn/bbsfiles/images/2014/12/201412190919_527879_2960432_3.png2:试样品反相色谱分析对波长的选择 同一个反相色谱柱,在柱温 流速 流动相配比相同而波长不同的条件下:分离度不同-----表现为波长越大,分离度越大;波长越小,分离度越小,基线噪音越大。灵敏度不同-----表现为波长越大,灵敏度越低;波长越小,灵敏度越高。试样的保留时间不随波长的改变发生变化。 说明:1:在反相液相色谱分析中,分离度大小与反相色谱柱的性能 柱温 流速 流动相的性质和配比有关系,与试样品的性质有关系。试样各组分在通过色谱柱而没有通过紫外检测器之前,保留时间以及分离度的大小就已经确定了,只是在通过紫外检测器时,由于紫外检测器波长发生了变化,紫外检测器的灵敏度(即单位浓度或单位质量的试样品通过紫外检测器检测到的信号强度)发生了变化,致使试样品各组分的峰高 峰面积发生变化,在表现形式上表现为分离度不同 灵敏度不同。2:尽管波长发生了不同的变化,但是,各组分的保留时间没有发生变化。这充分说明了各组分被洗脱的强度只与自身的性质(极性的强弱)以及色谱柱的性质(反相色谱柱)和柱温 流速 流动相的配比(流动相的极性)有关系。3:同种物质在不同的波长下紫外吸收不同,最大紫外吸收的波长为这种物质的截止波长,在反相液相色谱分析中,波长的设置都是流动相没有紫外吸收的波长,流动相如果有紫外吸收就会造成基线噪音和基线漂移。4:规定的范围内,波长越大,灵敏度越低,可以理解为:由于波长的增大,被测物的紫外吸收减弱,当被测物通过紫外检测器时,紫外检测器检测到的被测物的信号强度减弱,表现为峰高变低,峰面积变小,灵敏度降低。http://ng1.17img.cn/bbsfiles/images/2014/12/201412140931_527108_2960432_3.pnghttp://ng1.17img.cn/bbsfiles/images/2014/12/201412140931_527109_2960432_3.pnghttp://ng1.17img.cn/bbsfiles/images/2014/12/201412140932_527110_2960432_3.pnghttp://ng1.17img.cn/bbsfiles/images/2014/12/201412140954_527112_2960432_3.png3:试样品反相液相色谱分析对流动相配比浓度的选择 同一个反相色谱柱,在柱温 波长 流速相同,而流动相配比浓度(极性大小)不同的条件下:分离度不同(极性较小的试样)-----表现为流动相浓度增大(流动相极性减小),分离度降低,试样(极性较小的试样)保留时间缩短;流动相浓度减小(流动相极性增大),分离度增大,试样(极性较小的试样)保留时间延长。灵敏度不同(极性较小的试样)-----表现为流动相浓度增大(流动相极性减小),灵敏度增大;流动相浓度减小(流动相的极性增大),灵敏度减小。 说明:在反相液相色谱分析中,流动相浓度的改变就会造成流动相极性的改变。流动相浓度降低,流动相的极性增强,在反相色谱柱中的洗脱能力减弱------极性较大的试样先被洗脱,出峰时间早,保留时间变小;极性较小的试样后被洗脱,出峰时间晚,保留时间变大。流动相浓度增大,流动相的极性减小,在反相色谱柱中的洗脱能力增强-----极性大的试样后被洗脱,出峰时间晚,保留时间变大;极性较小的试样先被洗脱,出峰时间早,保留时间变小。http://ng1.17img.cn/bbsfiles/images/2014/12/201412140931_527108_2960432_3.pnghttp://ng1.17img.cn/bbsfiles/images/2014/12/201412141009_527114_2960432_3.png4:试样品反相液相色谱分析对流速的选择 同一个反相色谱柱,在柱温 波长 流动相配比相同而流速不同的条件下:分离度不同(极性较小的试样)-----表现为流速增加,分离度减小,试样保留时间减小;流速减小,分离度增加,试样保留时间增大。灵敏度不同(不明显)-------表现为流速增大,灵敏度减小;流速减小,灵敏度增大。http://ng1.17img.cn/bbsfiles/images/2014/12/201412141028_527118_2960432_3.pnghttp://ng1.17img.cn/bbsfiles/images/2014/12/201412141028_527120_2960432_3.png
[color=#666666][font=宋体][back=white][url=https://insevent.instrument.com.cn/t/5p]液相色谱[/url]([/back][/font][font='Times New Roman',serif][back=white]LC[/back][/font][font=宋体][back=white])方法开发是一个涉及多个步骤的过程,旨在确保分析结果的准确性和可靠性。以下是[url=https://insevent.instrument.com.cn/t/5p]液相色谱[/url]方法开发的一般流程:[/back][/font][font='Times New Roman',serif][back=white]1.[/back][/font][font=宋体][back=white]了解分析物的理化性质:在为分析物制定分析方法时,了解其理化性质至关重要。考虑的因素包括分析物的[/back][/font][font='Times New Roman',serif][back=white]pH[/back][/font][font=宋体][back=white]值、极性、溶解度和[/back][/font][font='Times New Roman',serif][back=white]pKa[/back][/font][font=宋体][back=white]。这些参数在[/back][/font][font='Times New Roman',serif][back=white]HPLC[/back][/font][font=宋体][back=white]方法开发中至关重要,影响着溶剂的选择和整个方法的成功与否。[/back][/font][font='Times New Roman',serif][back=white]2.[/back][/font][font=宋体][back=white]色谱条件优化:包括检测器、[/back][/font][font=宋体][back=white]色谱柱和[/back][/font][font=宋体][back=white]流动相的选择,并生成样品的“初始”色谱图。决定是开发[/back][/font][font='MS Gothic'][back=white]?[/back][/font][font=宋体][back=white]梯度方法还是等度方法,这两种方法都具有不同的优势,具体取决于分离要求和分析物的性质。[/back][/font][font='Times New Roman',serif][back=white]3.[/back][/font][font=宋体][back=white]色谱柱的选择:色谱柱是色谱仪的核心,在实现可靠、准确的分析中起着举足轻重的作用。选择好的色谱柱可确保良好的色谱分离效果,从而获得值得信赖的结果。反之,色谱柱选择不当会导致分离不充分和混乱,使结果无效或难以解释。[/back][/font][font='Times New Roman',serif][back=white]4.[/back][/font][font=宋体][back=white]流动相的选择:流动相的选择对于[url=https://insevent.instrument.com.cn/t/5p]液相色谱[/url]方法的开发至关重要。一般来说,反相[/back][/font][font='Times New Roman',serif][back=white]LC[/back][/font][font=宋体][back=white]的流动相包括水和作为改性剂的乙腈或甲醇。改性剂还有四氢呋喃[/back][/font][font='Times New Roman',serif][back=white](THF)[/back][/font][font=宋体][back=white]和异丙醇。反相色谱中流动相中水相的[/back][/font][font='Times New Roman',serif][back=white]pH[/back][/font][font=宋体][back=white]和离子强度在开发对条件微小变化不敏感的耐用方法中非常重要。[/back][/font][font='Times New Roman',serif][back=white]5.[/back][/font][font=宋体][back=white]方法验证:对最终的方法进行验证,检查它是否满足预期的分离效果与分析目的。这包括启动阶段的信息搜集、方法使用的目的明确、以及通过实例展示系统策略来进行方法筛选。[/back][/font][font='Times New Roman',serif][back=white]6.[/back][/font][font=宋体][back=white]优化和改进:根据初始实验结果,可能需要调整色谱条件以优化分离效果。这可能包括改变流动相的比例、调整[/back][/font][font='Times New Roman',serif][back=white]pH[/back][/font][font=宋体][back=white]值、改变温度等,以获得最佳的分离度和分辨率。[/back][/font][font='Times New Roman',serif][back=white]7.[/back][/font][font=宋体][back=white]样品制备:确保样品以适当的方式准备,以便在色谱分析中获得最佳结果。这可能包括样品的溶解、稀释、过滤等步骤。[/back][/font][/color]
在液相色谱中产品峰哈杂质峰分不开,怎么办?有哪些因素影响分离度?
农产品中氨基甲酸酯类农药检测的液相色谱条件优化研究曾 艳摘要:本文在行业标准《NY/761-2008蔬菜和水果中有机磷、有机氯、拟除虫菊酯和氨基甲酸酯类农药多残留的测定》用高效液相色谱法测定农产品中氨基甲酸酯类农药的基础上对色谱条件进行了优化研究。通过更换流动相及比例,建立了更高效的检测10种氨基甲酸酯类农药条件。研究表明,优化色谱条件后,色谱图的基线更平稳,组分保留时间平均缩短3.54min;在0.05-0.5mg/L范围内线性良好,相关系数为1.0(除涕灭威亚砜为0.9975外),检出限为0.001-0.007 mg/Kg;组分响应大大提高,峰高是之前的1.65-4.14倍,峰面积是1.62-3.97倍,连续7次进样的保留时间、峰高、峰面积相对标准偏差比优化前均有降低。优化的检测方法,快速简便,灵敏度高,准确可靠,有效的提高了工作效率,能满足农产品中氨基甲酸酯类农药的液相检测要求。关键词:甲醇,乙腈,高效液相色谱,氨基甲酸酯类农药Optimization of the method of HPLC todeterminated Carbamate pesticide in agricultural products residuesZENG Yan Abstract:This research was based onthe《NY/T 761-2008》byHPLC method to determination of carbamate pesticides in agriculturalproducts. This study was developed for the optimized chromatographic conditionsby changing the mobile phases and proportions, established a more efficientdetection of 10 kinds of carbamate pesticides. Study shows that the optimizedchromatographic conditions, the chromatograms baseline more smoothly, componentaverage save the retention times of 3.54 mins. The proposed methods showed agood linearity in the range of 0.05~0.5mg/Kg, with the linear correlationcoefficients of 1.0 (except aldicarb sulfoxid0.9975 ) and limits of detection of 0.001-0.007 mg/Kg . Components of responses are greatlyincreased: the peak heights are 1.65-4.14 times that of before and the peakareas are 1.62-3.97 times, 7 consecutive samples of retention times, peakheights and peak areas RSDs were lower than before optimization. The method is rapid, high sensitivity,accurate, reliable and effective and can be used for determination carbamate pesticidesin agricultural products.Keywords: Alcohol, Acetonitrile,HPLC, Carbamate pesticide,相对于有机磷杀虫剂, 氨基甲酸酯类农药以其残效小、选择性强、对人畜毒性较低等特点而被国内外广泛应用于病虫害的防治, 但不合理的使用仍对生态环境和农产品产生了一定的影响,随着国家对食品安全的重视不断提升,以及《农产品质量安全法》的颁布,果蔬中的氨基甲酸酯类农药残留检测至关重要。目前,资料报道的蔬菜、水果中氨基甲酸酯类农药残留检测方法主要有气相色谱法(GC)、气相色谱-质谱法(GC-MS)、 高效液相色谱法(HPLC)和液相色谱-质谱法(LC-MS)等。现行行业标准《NY/T761-2008蔬菜和水果中有机磷、有机氯、拟除虫菊酯和氨基甲酸酯类农药多残留的测定》即是用的高效液相色谱法(HPLC)。利用高效液相色谱检测果蔬、食用菌等农产品样本的原则是要尽可能在较短的时间内使混合物完全的分离和定量。对于多农残的痕量分析还要求有较高的灵敏度,因此最佳的色谱条件对于待测组分进行定性和定量分析尤为必要。本试验采用高效液相色谱仪对蔬菜中10种氨基甲酸酯类进行检测,通过对色谱条件进行优化研究,对检测结果进行比较,得到了比前报道和行业标准更为准确高效、灵敏度更高的检测方法,提高了农产品中氨基甲酸酯类农药的检测效率。1 材料和方法1.1供试材料乙腈、甲醇为色谱级(美国天地公司),柱后衍生试剂为pickering公司提供(包含邻苯二甲醛,OPA,巯基乙醇、OPA稀释剂和氢氧化钠溶液),水为超纯水,10种农药标准品(涕灭威亚砜、涕灭威砜、灭多威、三羟基克百威、涕灭威、速灭威、克百威、甲萘威、异丙威、仲丁威)购自农业部环境保护科研监测所(100μg/mL),纯度均大于99%,以乙腈稀释成合适浓度。供试样本为包含蔬菜、水果等农产品为试验样本,具体有菜豆,茄子,莴苣,黄瓜,结球甘蓝,油桃,马铃薯,黄果柑,随机选择。1.2 仪器 Agilent 1260高效液相色谱仪(四元泵),配荧光检测器(FLD),柱后衍生系统为美国科学系统公司的斯威特柱后衍生系统 双通道 PCR-2型,色谱柱为美国科学系统公司的Alltima C18 5μm4.6×250mm 色谱柱。1.3 实验方法1.3.1 标准液的配制将以上10种氨基甲酸酯类农药配制成0.005、0.05、0.1、0.2、0.5μg /mL5个浓度梯度的混合标准溶液,为了消除基质响应,在配制中加入基质溶液进行配制。1.3.2样品前处理样品前处理按《NY/T 761-2008蔬菜和水果中有机磷、有机氯、拟除虫菊酯和氨基甲酸酯类农药多残留的测定法》中进行操作,将样本中加入0.05、0.1、0.2、0.5mg/Kg的混合标液,每个样本7个平行。1.3.3分离条件的选择有报道用乙腈-水、乙腈-乙酸溶液、甲醇-乙酸溶液和甲醇-水作为流动相进行氨基甲酸酯类的液相检测,结果显示:乙腈-乙酸铵溶液和甲醇-乙酸铵溶液体系对10种氨基农药的分离效果和离子化程度都不如乙腈-甲酸溶液。因此,本试验在前人的基础上提高了流动相中有机相的比列,考察了只用乙腈-水和甲醇-水作为流动相,同时调整了流动相的流速。将配制好的标准品,按从低到高的浓度上机检测,检测条件为:FLD激发波长λex 330nm,发射波长λem 465nm,柱温为42℃,进样量为20uL,柱后衍生系统NaOH溶液和OPA的流速为0.3mL/min,水解温度为100℃,衍生温度为室温。优化后的色谱洗脱程序如表1所示。表 01 优化的梯度洗脱程序Table1 Optimizationof the gradient elution conditions
提高分离能力和改进分离的选择性一直是分离科学中具有挑战性的研究工作,而高效液相色谱和高效毛细管电泳则是分离科学中最重要的两个工具。高效液相色谱由于具有较高的分离能力和很好的重现性,已成为生命科学、药物科学等领域不可缺少的分析手段。但是,近十年来,高效毛细管电泳以其分离效率高、分析速度快、所需样品少以及消耗低等优势,展示了其在分析领域中巨大的应用潜力,已成为高效液相色谱的有力补充和竞争对手。 本论文即选择高效毛细管电泳和高效液相色谱这两种旨在提高分离能力的工具,对中草药山豆根中的有效成分和乌拉尔甘草叶中的营养物质进行了优化分离。此外,将高效液相色谱也成功地应用于敦煌壁画的颜料分析。 本论文共由以下五章组成: 第一章 对高效毛细管电泳的理论、技术以及在中草药分析方面的最新应用进行了综述,共引用了142篇参考文献。 第二章以0.04M含50%甲醇的柠檬酸(PH=4.5)作为缓冲溶液,在电压为25kV、检测波长为208nm的条件下,用毛细管区带电泳(CZE)方式对山豆根中的有效成分进行了分离实验。在这种优化的条件下,其时间和峰面积的精密度分别为1.39%-2.14...[img]http://www.instrument.com.cn/bbs/images/affix.gif[/img][url=http://www.instrument.com.cn/bbs/download.asp?ID=31318]毛细管电泳和高效液相色谱在中药分析中的应用[/url]
我想用液相色谱分离正构烷烃,并且把收集到的正构烷烃用馏分收集器收集,具体的方法是什么,用哪一种色谱柱,希望能够提供帮助!如果有相关资料,麻烦发一份邮件,拜托。







 我要推广仪器
我要推广仪器
 下载APP
下载APP