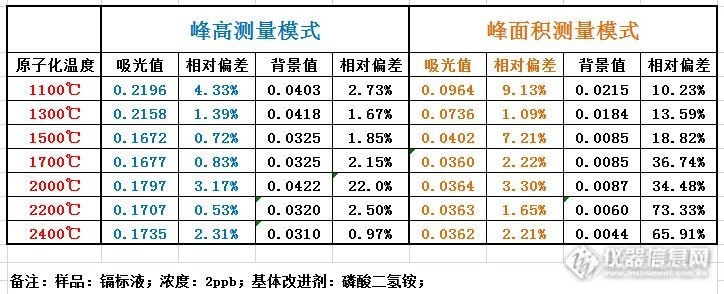
看到本版冰山版友关于石墨炉测镉的温度梯度试验http://bbs.instrument.com.cn/shtml/20140117/5160468/,感到很有意思;于是我利用今天下午半天的时间,用石墨炉对镉的单标液(2ppb)进行了一个原子化温度梯度的试验。一方面重新演绎一遍冰山版友的试验,另一方面也想通过我的试验,能给大家提供一个有趣味的话题。仪器:ZA3000型背景扣除方式:塞曼升温程序:干燥:80°~140° 30秒;灰化:400°~400° 15秒;原子化:1100°~2400°之间改变 5秒;清除:2000°~2500°。石墨管:C型高阻石墨管样品:Cd标液,浓度2PPb,进样量 20微升,每种测量模式重复三次测量。基体改进剂:磷酸二氢铵(2%),进样量5微升。http://ng1.17img.cn/bbsfiles/images/2014/01/201401231636_488710_1602290_3.jpg分析提示:(1)采用峰高测量模式,1500°和1700°是最佳结果,相对标准偏差最小;所以这也就是文献推荐的温度的出处。(2)峰高模式下,不同的原子化温度所得到的吸光值并不是一味地下滑,而是呈现U形。(3)采用峰面积模式吸光值也不是一味地下滑,在1700度~2400度之间基本保持不变,呈现L形。讨论问题:为何采用不同的测量模式所得到的吸光值的变化趋势不一致?

http://ng1.17img.cn/bbsfiles/images/2011/05/201105231514_295682_1621344_3.jpghttp://ng1.17img.cn/bbsfiles/images/2011/05/201105231515_295684_1621344_3.jpg日立横向与纵向加热石墨炉原子化瞬间的图片,看看图,单位距离上横向加热的温度梯度更大点!样品是加在石墨管正中心的,一般也就加20微升左右,样品在管子底部铺开会有一定面积,因为管子是桶形的,纵向铺开的长度会比横向大点儿,但纵向加热管在中心的温度梯度和横向管比要小,所以两种加热方式影响似乎应该差不多啊。
请教 高压梯度和低压梯度应用场合有什么不同? 具体地说,某一项分析如果采用高压梯度,是不是一定也可以采用低压梯度呢? 或者仅仅是经济条件方面不同呢?双泵的高压梯度和低压梯度仪器配置,资金相差大约有多少?
二元高压梯度(双泵+在线混合器)与四元低压梯度(单泵+低压梯度阀+脱气机+混合器),对于上面两个配置,现在哪个实用性更好?哪个洗脱效果更好?一般首选哪个配置? 顺便弱弱的问一下,如果我选二元高压梯度,对于像甲醇:水:冰醋酸(48:52:1)这种三种溶剂甚至四种的流动相该怎么去弄呢?它总共才二个溶剂通道吧。。。。
如题,请各位大神帮忙解答一下,高压梯度和低压梯度的优缺点各在哪里?高压梯度泵后混合可减少气泡的产生,但有人也说在检测器内(恢复常压后)可能会产生气泡。如果这样,是否高压梯度最好也配在线脱气?低压梯度的精度可以做到多少?对于串联泵来说,除了混合体积不够的原因外,比例阀切换频率如果和泵运行频率同步的话,是否也会造成基线不平(波浪形基线)?二元低压梯度最好的切换比例阀的方式是什么?按照时间周期或者是按照泵的运转周期?有没有熟悉waters或agilent的大神帮忙告知一下这两家是如何做的?感谢!
通常说的高压梯度和低压梯度到底有什么区别呢?一直很疑惑?
有什么参数可以预测等梯度或者梯度条件下的色谱保留时间呢?看到文献上有用 solute descriptors,比如 the excess molar refraction E (in cm3/10),the dipolarity/polarizability S, the solute’s effective hydrogen-bondacidity A and hydrogen-bond basicity B, and McGowan’s characteristicvolume V (in cm3 mol− 1/100).如果用这些参数来预测色谱保留时间的话,如何用软件计算得到这样的参数呢?或者还有没有其他的途径可以预测化合物的保留时间呢?
想做一个高压梯度泵梯度混合器的了解,请大家有二元高压梯度仪器的说说自己的仪器型号及梯度混合器的体积。
请教:进行梯度洗脱时,用低压梯度还是高压梯度好?有什么区别?谢谢!
大家好,我有一套岛津HPLC,工作站是LC-Solution,2个LC-10AD 泵组成高压梯度,但是是当我检查梯度准确性是,发现线性梯度的曲线是弯曲的,而我的梯度曲线值“curve” 是默认的“0”,2个泵流速是准确的,请问是不是梯度曲线的问题啊?改设置为多少呢?
怎样做梯度洗脱啊,是直接在泵上设置就行么?内梯度和外梯度的拆别在哪里?
用液相色谱测定TBHQ和BHT流动相: 1.5%(体积分数)乙酸-甲醇溶液(A),1.5 %(体积分数)乙酸—水溶液(B)洗脱梯度: 0min -5min ,流动相A为30%,5min-20min,流动相A从30%线性增至80%,20min -30min,流动相A为80% 梯度洗脱设置 Time Module Action Value0.01 pumps B.Conc 705.00 pumps B.Conc 7020.00 pumps B.Conc 2030.00 pumps B.Conc 2031.00 pumps B.Conc 7042.00 SCL stop 0

1 .梯度洗脱等于无限个等度洗脱http://ng1.17img.cn/bbsfiles/images/2015/12/201512131829_577712_3047134_3.jpgfigure 1a所示,条件是60%B的等度洗脱,我们会发现,在色谱的前端,峰并没有很好地分离,而在色谱的尾端,虽然峰分离的很好,但峰变的很宽。如果降低B的比例,前面的峰的可以得到分离,而后端的峰宽将会变的更宽;如果提高B的比例,后端的峰可以快速流出,峰宽也会窄一些,但前端的峰会更加难以分离,所以,遇到这种情况,很难用等度洗脱得到很好的分离。为什么会出现这种情况?通过计算,发现1a中样品的保留因子范围是0.934(34/0.9 ≈ 38),样品的极性差别很大,而通常等度洗脱的保留因子范围是120(20/1=20), 1a中保留因子的范围38远远大于20,所以是不适合用等度洗脱的。figure 1b,梯度洗脱的条件是 50-100% in 5 min,会发现不论是前端还是后端,峰都得到了很好的分离,并且峰宽几乎是一样的Figure 1c做了一个试验,将figure 1中分析的15种物质分成了5组,每组都是等度洗脱,B的比例分别是49%,61%,72%,80%和88%,每组的保留因子大概都是k等于3,峰宽也几乎一样,是不是和figure 1b很相似?如果将c中的等度洗脱的条件叠加,就是一个梯度洗脱,也就是说梯度洗脱可以理解为是有无数个比例变化很小的等度洗脱的叠加。2.梯度洗脱的窄峰宽和等峰宽梯度洗脱的最大好处就是能够窄的峰宽,并且同一梯度内的峰宽都是一样的,为什么呢?http://ng1.17img.cn/bbsfiles/images/2015/12/201512131830_577713_3047134_3.jpg如Figure 2b所示,梯度条件是 5-100% B ,梯度时间是20min,2a显示了梯度变化时,保留因子k(虚线)的变化以及峰宽(实线)的变化。在梯度开始时,有机相比例低,保留因子k最大,峰还停留在柱头,峰宽为0,随着有机相比例的增加,物质开始柱子内移动,有机相比例不断增加,k值也一直下降,物质在柱子中不断移动直到流出。峰1和峰2的保留时间虽然不一样,但其移动的模式是一样的,并且k的变化曲线也是一样的,所以在梯度洗脱条件下,其峰宽也是一样的。1梯度洗脱等于无限个等度洗脱figure 1a所示,条件是60%B的等度洗脱,我们会发现,在色谱的前端,峰并没有很好地分离,而在色谱的尾端,虽然峰分离的很好,但峰变的很宽。如果降低B的比例,前面的峰的可以得到分离,而后端的峰宽将会变的更宽;如果提高B的比例,后端的峰可以快速流出,峰宽也会窄一些,但前端的峰会更加难以分离,所以,遇到这种情况,很难用等度洗脱得到很好的分离。为什么会出现这种情况?通过计算,发现1a中样品的保留因子范围是0.934(34/0.9 ≈ 38),样品的极性差别很大,而通常等度洗脱的保留因子范围是120(20/1=20), 1a中保留因子的范围38远远大于20,所以是不适合用等度洗脱的。figure 1b,梯度洗脱的条件是 50-100% in 5 min,会发现不论是前端还是后端,峰都得到了很好的分离,并且峰宽几乎是一样的Figure 1c做了一个试验,将figure 1中分析的15种物质分成了5组,每组都是等度洗脱,B的比例分别是49%,61%,72%,80%和88%,每组的保留因子大概都是k等于3,峰宽也几乎一样,是不是和figure 1b很相似?如果将c中的等度洗脱的条件叠加,就是一个梯度洗脱,也就是说梯度洗脱可以理解为是有无数个比例变化很小的等度洗脱的叠加。2梯度洗脱的窄峰宽和等峰宽梯度洗脱的最大好处就是能够窄的峰宽,并且同一梯度内的峰宽都是一样的,为什么呢?如Figure 2b所示,梯度条件是 5-100% B ,梯度时间是20min,2a显示了梯度变化时,保留因子k(虚线)的变化以及峰宽(实线)的变化。在梯度开始时,有机相比例低,保留因子k最大,峰还停留在柱头,峰宽为0,随着有机相比例的增加,物质开始柱子内移动,有机相比例不断增加,k值也一直下降,物质在柱子中不断移动直到流出。峰1和峰2的保留时间虽然不一样,但其移动的模式是一样的,并且k的变化曲线也是一样的,所以在梯度洗脱条件下,其峰宽也是一样的。
卤乙酸的水溶液可以直接进HP-5色谱柱吗? 测定水中卤乙酸含量,可以用卤乙酸的标准品的水溶液做浓度梯度,绘制标准曲线吗? 问题补充: 使用的是ECD检测器。需要绘制卤乙酸的标准工作曲线。是卤乙酸用水溶,还是卤乙酸用甲醇做溶剂?能直接水溶卤乙酸进HP-5的色谱柱吗?
高压二元梯度与四元低压梯度究竟哪一个好呢,请大家踊跃发言
单梯度与双梯度磁场的比较
如题:梯度升温、梯度改变载气流速等如何应用?有实例么?
看到文献中这样的一个表达 : 流 动 相 为 石 油 醚-乙 酸 乙 酯 (100∶1~1∶10),收集洗脱液并合并,减压蒸干溶剂,得到 Frac.A1~A10馏分。想请问这里的100∶1~1∶10包括哪10个梯度啊?初出茅庐,还望大佬解惑!
液相梯度洗脱中,分线性梯度、凸形梯度、凹形梯度、分段梯度,请教一下,凸形和凹形的程序如何设置呢?这两种梯度是否可以采用分段梯度来达到同样的效果呢?
由于JJC 705-202规程中对于梯度误差的检定只给出了二元的,现在四元梯度使用越来越多,各家的检定都是自定一套,没有统一标准。如何定才最合理?很想知道大家的想法。
液相中有个问题,等度洗脱完全没有问题,基线ok,pressure fluctuation ok,rsd ok。走梯度的时候,两相到一定比例,会出现一巨大的峰,或者说那是一包吧。流动相用到乙腈,缓冲盐,缓冲盐是一定浓度溶解在乙腈中的,就是俩含不同浓度缓冲盐溶解在乙腈里做为流动相。检测波长是210nm,看了些资料,说的是走梯度的时候,就是会出鬼峰,什么情况都有。我想,必定是里面有东西,它才会有吸收, 而且还那么大一团。 不然基线也没法升上去呀? 说是流动相里面的东西聚集到一定程度就被洗脱下来,这就是鬼峰的来源。如果仪器硬件有故障会出现这样的故障吗? 我想是不会的,除非是脏。另外就是柱子了,如果柱子也没问题,那就只能是流动相了,我讲的对吗?
HPLC有等强度(isocratic)和梯度(gradient)洗脱两种方式。等度洗脱是在同一分析周期内流动相组成保持恒定,适合于组分数目较少,性质差别不大的样品。梯度洗脱是在一个分析周期内程序控制流动相的组成,如溶剂的极性、离子强度和pH值等,用于分析组分数目多、性质差异较大的复杂样品。采用梯度洗脱可以缩短分析时间,提高分离度,改善峰形,提高检测灵敏度,但是常常引起基线漂移和降低重现性。 梯度洗脱有两种实现方式:低压梯度(外梯度)和高压梯度(内梯度)。 两种溶剂组成的梯度洗脱可按任意程度混合,即有多种洗脱曲线:线性梯度、凹形梯度、凸形梯度和阶梯形梯度。线性梯度最常用,尤其适合于在反相柱上进行梯度洗脱。 在进行梯度洗脱时,由于多种溶剂混合,而且组成不断变化,因此带来一些特殊问题,必须充分重视: ①要注意溶剂的互溶性,不相混溶的溶剂不能用作梯度洗脱的流动相。有些溶剂在一定比例内混溶,超出范围后就不互溶,使用时更要引起注意。当有机溶剂和缓冲液混合时,还可能析出盐的晶体,尤其使用磷酸盐时需特别小心。 ②梯度洗脱所用的溶剂纯度要求更高,以保证良好的重现性。进行样品分析前必须进行空白梯度洗脱,以辨认溶剂杂质峰,因为弱溶剂中的杂质富集在色谱柱头后会被强溶剂洗脱下来。用于梯度洗脱的溶剂需彻底脱气,以防止混合时产生气泡。 ③混合溶剂的粘度常随组成而变化,因而在梯度洗脱时常出现压力的变化。例如甲醇和水粘度都较小,当二者以相近比例混合时粘度增大很多,此时的柱压大约是甲醇或水为流动相时的两倍。因此要注意防止梯度洗脱过程中压力超过输液泵或色谱柱能承受的最大压力。 ④每次梯度洗脱之后必须对色谱柱进行再生处理,使其恢复到初始状态。需让10~30倍柱容积的初始流动相流经色谱柱,使固定相与初始流动相达到完全平衡。
低压梯度比高压成本低很多,即使加上在线脱气装置,成本还是低,既然这样为什么又有人去买高压梯度洗脱呢?高压梯度应该有其特点,不知道在应用上与低压有什么不同,原理大家都懂。
小弟做游离氨基酸,先用的梯度是:0.01min B 0% ,12min B 15% ,30min B 34% ,30.01min B 100% ,40min B 0% ,50min B 0%.这是经典的做氨基酸的梯度,但我用得不行,峰总有几个连在一起已经排除了其他条件对峰的干扰可能(全部按标准做的)后认为应该提高洗脱能力,对于反向柱,降低B的含量于是采用这个梯度:0.01min B 0% ,13min B 7% ,23min B 23% ,29min B 35% ,35min B 40% ,40min B 100% ,45min B 100% ,47min B 0%这个梯度已经降低了B的含量,但还是无法把峰分开哪位大侠做过的指点一下偶的迷津,帮我设计一个合理的梯度在此多谢了买新柱子老板是绝对不会答应的
关于梯度淋洗,想知道四元梯度和二元梯度的区别,四元是低压的吗,现在还有低压淋洗的应用吗
关于梯度淋洗,想知道四元梯度和二元梯度的区别,四元是低压的吗,现在还有低压淋洗的应用吗
我做的是样品中的黄酮定性定量,试了许多文献的方法,只有芦丁出峰,但按理说应该还有槲皮素、山奈酚等等,一个是因为其他品种的样品有,另一个是总黄酮含量大小和芦丁含量大小不一致。在我尝试了浓缩、纯化、酸水解都不行之后,我开始摸索液相条件我看了社区的一些精华帖,选定了0-48min,5%-95%乙腈(另一个是1%乙酸),打算先测一下进样体积和流速的影响,结果发现在这个梯度下,无论怎么换别的,只有两分钟左右的溶剂峰,其余一个峰都没有。所以,我觉得可能得用分段的梯度,但是这个分段怎么分,每一段的流动相占比我不知道该怎么设置,于是就胡乱设置了一下。比如48min平分成六段,一段8分钟,让乙腈从5%开始往上升,升到100%,再走一个初始比例。我想问的是:1. 我上面的思路是对的吗?2. 大家知道梯度怎么分段设置吗?3. 有的方法混标跑的很好,槲皮素那些都能出峰,但是样品就是不出槲皮素的峰,这是不是说明我样品里就是没有这些东西?4. 我现在尝试分析别人的梯度,比如他们的槲皮素8分钟出峰,而梯度是0-10min,30-40%乙腈,这能不能说明,(40-30)/10*8+30=38,38%的乙腈可以洗脱槲皮素,我应该用38%左右的乙腈多走一会?
我用的可做4元梯度,我想问的是,在做多种组分时,怎样根据等度图谱,设梯度洗脱,可使基线不会有太显著飘逸和梯度变化的时间点以及梯度曲线怎样选择?
梯度洗脱所用的溶剂纯度要求更高,以保证良好的重现性。进行样品分析前必须进行空白梯度洗脱,以辨认溶剂杂质峰,因为弱溶剂中的杂质富集在色谱柱头后会被强溶剂洗脱下来。用于梯度洗脱的溶剂需彻底脱气,以防止混合时产生气泡。
[align=center]开发了一个梯度方法,当我在我的系统上运行它重复性很好,但我不能在另一个系统上得到相同的结果?[/align]当梯度法从一个高效液相色谱系统转移到另一个系统时,偶尔会出现一些困难。除非系统完全相同,否则用户通常会在保留时间上发生一些变化。大多数情况下,留存率的变化不会影响到解决方法的效果,人们通常可以忽略这些差异。另一方面,通过对潜在原因的正确理解,我们可以调整梯度,以从不同的HPLC系统获得相同的性能。梯度驻留体积:它是梯度混合点和柱顶之间的体积。在你通过注入样品开始分析后,梯度将不会到达柱的顶部,直到梯度停留体积被清除。这意味着你的样本最初要经历一段等稳迁移的时期,直到梯度赶上。由于不同系统的梯度停留体积不同,这种等稳偏移时间也会不同,可能导致滞留时间的差异,甚至影响分辨率。另一种可能是梯度本身。系统与系统之间可能存在组成差异。对于今天生产的大多数HPLC系统,这应该只是次要的问题。但是,一般来说,任何梯度系统都能在成分的中间范围,即50% a和50% B的混合物中,提供具有最高准确度的成分。当非常不成比例的a和B混合时,例如5% a或95% a,准确性就会受到影响。[align=center]哪些可能导致我的方法传输问题?[/align]最简单的事情是比较你的梯度两个系统。为了做到这一点,你断开色谱柱,并在梯度的b溶剂中添加紫外线吸收剂。如果你使用的是反相体系,你可以在b溶剂中加入10mg /L的对羟基苯甲酸丙酯。然后在两个系统上运行渐变并记录基线。然后将这两个情节相互比较。你需要找到梯度开始的点,你还需要测量梯度剖面。如果你的梯度是线性的,那么你只需要检查梯度的斜率。很有可能你会发现两种仪器之间的梯度开始是不同的,而轮廓是非常相似的,只是被一些时间抵消了。在这种情况下,你有一个不同的梯度停留体积。[align=center]是否有一种简单的方法来补偿驻留体积的差异?[/align]有一个解决方案大多数时候都是有效的。如果梯度停留体积在你的方法转移到的系统上较小,你可以通过在梯度开始时设计一个补偿体积差异的等稳部分来补偿停留体积的不足。剩下的梯度保持不变。另一方面,如果第二个系统的梯度停留体积比第一个系统的梯度停留体积大,则情况更困难。原则上,您可以启动梯度,然后在延迟一段时间后注入样品,这段时间可以解释两种系统之间梯度停留体积的差异。但在自动系统上,这可能是不可能的,因为注入会触发梯度的开始。在这种情况下,您可能需要回到原点并重新开发该方法。[align=center]我能做什么来防止这种情况在未来发生?[/align]您可以通过为最终将使用该方法的HPLC系统开发该方法来避免这种情况。这需要一些远见和一些计划,但通常不是不可能的。您首先需要做的是描述可能用于您的方法的系统。对于每个系统,你需要知道两件基本的事情:梯度驻留体积和合成精度。你可以在一个实验中得到这两个信息:如上所述,你在你的b溶剂中添加一个紫外线吸收剂。然后你在0% B到100% B的增量5%的多个步骤梯度。流量应该是通常使用的流量,所以最有可能你将使用1毫升/分钟的流量。步骤之间的间隔应该是几分钟,或者5分钟。现在在不同的系统上运行这个梯度,而不需要一个柱,并记录检测器的响应。为每一步编程的时间和实际发生的步骤之间的时间延迟给你梯度延迟时间。台阶的高度给了你构图的一个度量。步骤会被抹去一点,这是系统中混合体积的函数。现在您已经确定了系统的特征,您可以围绕系统的特征设计您的方法。正如我之前提到的,最大的问题通常是渐变驻留体积。如果您知道需要转移您的方法的系统比开发您的方法的系统有更大的驻留体积,那么您应该在您的方法开发中自动添加一个等稳步骤,以补偿这种差异。如果目标系统的驻留体积小于开发系统的驻留体积,那么您应该能够通过在转移方法时在方法的开始部分添加一个梯度延迟时间来简单地补偿这一点。如果在渐变过程中存在组成差异,你也可以通过调整渐变轮廓来弥补这些差异,但我从来没有遇到过这种情况。本讨论假设柱与起始流动相处于完全平衡。我偶尔会遇到这样一种情况,在常规分析中,梯度彼此跟随得如此之快,以至于柱在起始流动阶段永远不会恢复平衡。你会发现,如果你的第一个梯度总是给出不同于后续梯度的结果,你就会遇到这种情况。这可能有利于加快方法的速度,但当将该方法转移到具有不同驻留体积的另一个系统时,这可能会造成困难。







 我要推广仪器
我要推广仪器
 下载APP
下载APP