假设在[url=https://insevent.instrument.com.cn/t/Mp]气相色谱仪[/url](FID)上分析样品,用面积归一法得到结果,组分A=30%、B=70%,请问该结果代表的是质量含量还是体积含量?
请问一般来说0.06%高氯酸是质量百分含量还是体积含量?我们领回来的是分析纯的高氯酸,质量含量是70-75%,是不是还要进行换算?
安捷伦GCMS序列中输入样品质量和定容体积后,数据处理的时候计算结果是错误的,不能像岛津GCMS那样,直接输入样品质量和定容体积,然后数据出来就是样品中最终含量。另外安捷伦GCMS序列中的体积是进样体积不是定容体积,只能输入稀释,数据处理的时候,浓度那列显示的是百分含量。可否通过“用户自定义”来设置公式进行自动计算呢?求大神指教。
样品是从车上取得,用气袋去的液体然后挥发成气体。主要是原料裂解碳四和醚后碳四。需要分析其中的含氧化合物甲醇,二甲醚和MTBE的含量。用的是外标法,进入色谱是气体样品。标气是买的标准气体,标气此时应该选用质量比还是体积比?有分析类似的样品的同行或是相关的老师能解答一下。
我们的气相色谱测定的样品都是溶解在溶剂中的,但是要怎么测定某个气体样品的含量呢?谢谢!
[url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]中所显示的含量是什么含量?是氢焰检测器。原来好多人都说是质量含量,但是今天一个色谱公司的说是摩尔含量,我顿时晕倒。到底是什么含量呀?谢谢!
超临界流体萃取法——毛细管[url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]法分析牡丹皮及制剂中丹皮酚的含量第二军医大学长征医院药材科 (上海 200003) 缪海均 柳正良* 李云华* 邵元福*第二军医大学药学院[B]摘要[/B] 本文采用超临界流体萃取法(supercritical fluid extraction,SFE)提取中药牡丹皮及其成方制剂中丹皮酚,以氯仿作改性剂,在温度90℃,压力4000 psi下,二氧化碳动态萃取体积3 ml 静脉萃取时间5 min为最佳。该法简便快速,萃取完全。用大孔径毛细管柱[url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]法作含量监测,结果:相关性好(r=0.9999),中药与制剂的回收率分别为97.8%,RSD=2.35%(n=3);100.3%,RSD=1.89%(n=3)。结果准确,灵敏,分辨率好。为中药有效成分的提取和质量控制提供了有效可靠的方法。[B]关键词[/B] 超临界流体萃取法; 牡丹皮; 丹皮酚; 毛细管柱[url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]法中药的成分极其复杂,在定量分析中,样品的提取是非常关键的,超临界流体萃取是近代分离领域中出现的先进的样品制备技术,将传统的蒸馏和有机溶剂萃取结合在一起,利用超临界流体的优良溶解能力,达到分离、纯化的目的。丹皮酚(paeonol)是毛茛科植物牡丹(Paeonia suffruticosa Andr)的主要挥发性有效成分,具有祛风镇痛、降压、止血、抗炎症、抗菌、抑制血小板凝集等多种药理作用 [1] 。本文用超临界二氧化碳流体对牡丹皮及其成方制剂中丹皮酚的萃取条件进行了系统的研究,筛选出压力、温度、CO 2动态萃取量、静态萃取时间和改性剂加入量等五变量的最佳条件,用大孔径毛细管柱[url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]测定了丹皮酚的含量,其结果准确、灵敏,分辨率好。[B]1 实验部分[/B]仪器、试剂和药品 仪器:100DX、100DM注射泵,SFX210超临界萃取器(美国ISCO公司),HP5890series II[url=https://insevent.instrument.com.cn/t/Mp]气相色谱仪[/url](美国HP公司),NEC Pewermate 325(日本)。试剂:二氧化碳(99%)(上海酒精总厂),其余试剂均为分析纯。药品:丹皮酚标准品(中国药品生物制品检定所),药材及成方制剂购于长海医院。
心力丸是本所科研成果,药厂注册品种,国家中药保护品种,质量标准收载于卫生部中药成方制剂第十七册(WS3—B—3150—98)。本品具温阳益气、活血化瘀功能,用于心阳不振、气滞血瘀所致胸痹心痛、胸闷气短、心悸怔忡等证及冠心病、心绞痛。该制剂是由麝香、人参、附子、红花、人工牛黄、蟾酥等中药制成的浓缩丸,现国家规定处方中麝香以人工麝香等量替代使用,因此,人工麝香中主要有效成分麝香酮的含量[1],对控制该产品质量和保证临床用药安全有效具有重要意义。已有对六神丸等制剂中麝香酮进行测定的文献[2-5]报道。本文用[url=https://insevent.instrument.com.cn/t/Mp][color=#3333ff]气相色谱[/color][/url]法测定心力丸麝香酮的含量,方法简便、准确,可以有效控制该产品质量。1 仪器和试剂1.1 仪器岛津GC17A[url=https://insevent.instrument.com.cn/t/Mp][color=#3333ff]气相色谱仪[/color][/url],FID 检测器,N2000数据工作站(浙江智达) METTLER AE240电子分析天平 AS5150A超声波仪。1.2 试剂、试药麝香酮对照品(中国药品生物制品检定所,批号:071920006,含量测定用,质量分数99.8%) 心力丸(广东省药物研究所制药厂) 无水乙醇、乙醚等试剂均为分析纯 压缩空气、H2、N2均为高纯度的气体(广州气体厂,质量分数达99.9%以上)。2 方法与结果2.1 色谱条件毛细管柱:SPBTM1 FUSED SILICA Capillary Column(30 m×0.32 mm×1.0 μm) 柱温:200 ℃ 进样口温度:210 ℃ 检测器温度:225 ℃ 载气:N2 流速:2 mL/min 柱前压:80 kPa 空气:300 mL 氢气:30 mL 尾吹:30 mL 分流:50∶1 进样量:1 μL。在上述条件下,供试品溶液中麝香酮色谱峰能达到基线分离,供试品与邻峰的分离度大于2.0,峰形对称,人工麝香阴性供试品无干扰,方法具有专属性,按麝香酮计算理论塔板数不低于4 500。色谱图见图1-图3。图1 麝香酮对照品的[url=https://insevent.instrument.com.cn/t/Mp][color=#3333ff]气相色谱[/color][/url]图(M.麝香酮)(略)Figure 1 Gas chromatograms of muscone图2 心力丸供试品的[url=https://insevent.instrument.com.cn/t/Mp][color=#3333ff]气相色谱[/color][/url]图(M.麝香酮)(略)Figure 2 Gas chromatograms of Xinli pills图3 人工麝香阴性[url=https://insevent.instrument.com.cn/t/Mp][color=#3333ff]气相色谱[/color][/url]图(略)Figure 3 Gas chromatograms of negative sample2.2 线性关系考察精密称取麝香酮对照品0.151 94 g,置100 mL量瓶中,加无水乙醇使溶解并稀释至刻度,摇匀 再分别精密量取0.5、1、2、4、5、6、8、10 mL置10 mL量瓶中,加无水乙醇至刻度,摇匀,精密吸取1 μL进样,测定峰面积,以对照品质量浓度(ρ)为横坐标,峰面积(A)为纵坐标,求得回归方程为A=291382. 2ρ+18117.0,r=0.999 6,麝香酮线性范围为0.076~1.5 mgmL-1。2.3 供试品溶液制备取本品,研细,精密称取3.5 g,置具塞锥形瓶中,加入乙醚30 mL,密塞,冷浸12 h,滤过,残渣和具皿用少量乙醚洗涤数次,合并乙醚液,挥去乙醚,残渣加无水乙醇使溶解并定容至5 mL,摇匀,用0. 45 μm滤膜滤过,取续滤液,即得。2.4 测定方法精密吸取供试品溶液和对照品溶液1 μL,分别注入[url=https://insevent.instrument.com.cn/t/Mp][color=#3333ff]气相色谱仪[/color][/url]中,记录色谱图,以外标法计算供试品中麝香酮含量。2.5 稳定性试验取同一供试品溶液制备后室温放置,分别在0、1、2、4、6、8、10 h进样测定,每次进样1 μL,测得麝香酮平均含量490.4 μg/g,RSD为0.29%。结果表明供试品在10 h内测定结果稳定。2.6 精密度试验取麝香酮对照品溶液(0.759 7 mg/mL)重复测定6次,测得麝香酮峰面积的RSD值为0.15%。2.7 重现性试验取同一批供试品,平行6次精密称取,按“2.3”项下方法制备供试品溶液,依“2.4”项测定,计算供试品中麝香酮含量,结果其RSD值为1.4%,表明方法重现性好。2.8 回收率试验精密称取已知含量的心力丸(麝香酮含量495. 0 μg/g)共9份,置具塞锥形瓶中 取高(1.5194 mg/mL)、中(0.759 7 mg/mL)、低(0.075 97 mg/mL)3个浓度的对照品溶液各3份,每份2 mL,置蒸发皿中,通风柜中挥干乙醇,残渣用适量乙醚溶解,移入称取有供试品的具塞锥形瓶,用乙醚洗涤具皿数次,将洗涤液并入锥形瓶,乙醚用量为30 mL,其余按“2.3”方法制备,按“2.4”方法测定,结果见表1。2.9 样品测定取本品6批,按“2.3”项下方法制备供试品溶液,按“2.4”法进行测定,结果见表2。表1 麝香酮回收率试验结果(略)Table 1 Recovery test of muscone表2 样品测定结果(略)Table 2 Assay test of samples3 讨论3.1 提取方法的选择分别考察了超声提取法和浸渍法,结果见表3。结果表明:用乙醚作溶剂的2种提取方法有偏差(相对偏差为3.1%),浸渍法提取比超声法提取损失少,或者说提取较完全 在产品含量测定时采用浸渍法提取,提取适宜时间为12 h 在生产过程中的产品质量检测则可采用超声提取方法。3.2 色谱柱的选择曾采用强极性毛细管柱(SHIMADZU CBP20睲25025)进行试验,结果麝香酮很难达基线分离,且被测组分峰有拖尾现象。表3 两种提取方法麝香酮含量测定结果(略)Table 3 Muscone contents from two batches of extractions【参考文献】[1] 唐洪梅,黄樱华,李得堂,等.[url=https://insevent.instrument.com.cn/t/Mp][color=#3333ff]气相色谱[/color][/url]法测定人工麝香中麝香酮的含量[J].中国实验方剂学杂志,2007,13(9):4-6.[2] 林巧玲,卓婷.[url=https://insevent.instrument.com.cn/t/Mp][color=#3333ff]气相色谱[/color][/url]法测定保婴散中麝香酮的含量[J].广东药学院学报, 2006,22(3):257-258.[3] 袁劲松,汤翠娥.[url=https://insevent.instrument.com.cn/t/Mp][color=#3333ff]气相色谱[/color][/url]法测定紫金胶囊中麝香酮含量[J].中国医院药学杂志, 2003,23(2):89-91.[4] 邹巧根,苏建俊.[url=https://insevent.instrument.com.cn/t/Mp][color=#3333ff]气相色谱[/color][/url]法测定麝香保心丸中麝香酮的含量[J].中国中药杂志, 1994,19(7):418-420.[5] 王强,毕开顺,陈金泉,等.六神丸中麝香酮含量的GC法测定[J].中国药科大学学报, 1995,25(6):87-89.
摘要:讨论了用气相色谱内标法测定合成树脂乳液中未反应完的残余单体含量的方法,并对该方法的精密度、准确度进行了考察/实验表明,该方法简单易行,准确可靠,适用于各种合成树脂乳液样品。 0.引言 自国家十项强制性标准颁布以后,市面上各种涂料中的有害物质必须达到国家相关限量标准。由于合成树脂乳液中未反应的残余单体对人的身体健康和环境会带来不同程度的影响,为此必须设法控制乳液中残余单体的浓度……..目前国科多采用顶空进样技术分析乳液中的残余单体含量,由于仪器投资较大,且样品回收率也不理想,样品前处理较烦锁,对于中小企业这样投资较少,易于在中小企业中推广,该分析方法较难于推广,而我们介绍的是普通分析方法。采用小口径毛细管柱,用氢火焰离子化检测器(FID)进行检测,以内标法定量。柱温采用程序升温,分离效果十分理想。其加标回收率分别在94%-103%之间,分析结果的精密度、准确度完全达到检测要求。 1.1实验部分:1.1仪器和试剂电子分析天平(万分之一);GC7800气相色谱仪(带有分流装置);1.0ul微量进样器;20ml带胶塞小玻璃瓶若干;医用注射器1ml、2ml各两只;小口径毛细柱DB-17HT(0.25mmx30mx0.15um;最高使用温度为360℃);积分仪或色谱工作站。醋酸乙烯酯VAM(色谱纯)、甲基丙烯酸甲酯MMA(色谱纯)、苯乙烯ST(色谱纯)、丙烯酸丁酯BA(色谱纯)丙烯异辛酯2-EHA(色谱纯)、丙酮(分析纯)配成4 1混合水溶液(作稀释剂),水(纯净水或蒸馏水)。内标物:环已酮(色谱纯) 1.2测定原理 试样中加适量内标物并用少许丙酮(4 1)稀释摇匀后,用微量注射器将稀释后的溶液注入气相色谱仪,样品被载气带入色谱柱,在柱内被分离成相应的组份,用氢火焰离子化检测器检测并记录色谱图,反数据用内标法计算试样溶液中各待测残余单体的含量。 1.3测定条件以样品中各组分是否完全分离开为依据而确认测定条件: 气化温度:280℃检测温度:320℃载气:氮气:纯度≥99.99%,变色硅胶 5A分子筛除水、除油,柱前压力为60Kpa(30℃);氢气:纯度≥99.99%,变色硅胶 5A分子筛除水、除油,柱前压力为65Kpa(30℃);空气:变色硅胶 5A分子筛除水、除油,柱前压力为55Kpa(30℃);柱温采用程序升温:初温30℃,恒温3min,以10℃/mm升温速率升至140℃,再以50℃/min升温速率升至260℃保持4min.分流比:45:1进样量:0.2ul1.4相对校正因子的测定1.4.1标准样品的配制于20ml样品瓶中分别称取0.03g(精确至0.0001g)的醋梭乙烯酯VAM、丙烯酸丁酯Bt等待测单体的色谱纯品和内标物环已酮,加入纺2mL丙酮(4 1)稀释,用力摇匀3min.(注意:每次衡量后应立即将样品瓶盖紧,以防止样品挥发损失。) 1.4相对校正因子的测定待仪器稳定后,吸取0.2uL标准样品注入色谱仪,记录色谱图和色谱数据……。典型合成树脂乳液的色谱图各单体及内标物的相对保留时间分别是:VAM1.771’;MMA3.078’;BA5.498’;St5.683’;2-EHA8.495’;内标环已酮6.218’1.4.3相对校正因子的计算醋酸乙烯酯、丙烯酸丁酯、苯乙烯等各需待测的残余单体对环已酮的相对校正因子Fi按下式计算:式中:mi——待测残余单体各自的质量;gAs——内标物环已酮的峰面积;ms——内标物环已酮的质量;gAi——待没残余单体各自的峰面积。 1.5连续平行测得待测残余单体各自对环已酮(内标物)的相对校正因子Fi的平等偏差应小于0.05.1.5加标回收率试验为了证明测定结果的可靠性,称取一定量的各待测单体纯品配制一已知尝试溶液,按上述分析方法测定各溶液的浓度,计算各自的回收率,结果见表(1) 1.6样品的测定将样品搅拌均匀后,在20mL样品瓶中准确称取2g左右和一小滴(用1mL注射器)内标物(约0.001g)环已酮于样品瓶中,加入适量(2~3mL)的丙酮(4 1)稀释剂,立即加盖瓶塞,充分摇匀2min.在相同于测定校正因子的分析条件下,用1ul微量进样器取0.2ul该试液注入色谱仪,并记录色谱图和数据,根据待测残余单体各自对内标物的保留时间进行定性。 1.7结果的计算待测残余单体各自的质量分数按下式计算:式中:Fi——待没单体各自对内标物的相对校正因子;Ms——内标物的质量,gAi——试样中待测残余单体各自的峰存积;Mi——待没试样的质量,gAs——内标物的峰面积取平行测定2次结果的算术平均值作为试样中各待测单体的测定结果。 1.8重现性同一操作作者两次测定结果的相对偏差小于0.5%2.结果与讨论(1)该分析方法选用中等极性小口径耐高温的毛细柱,进样量不能大于0.2ul,且分流比要足够大。由于各残余单体的沸点相对较高,所以我们选用耐高温的毛细柱,且该柱对酯类有较佳的分离效果。 (2)汽化室内须配有石英玻璃衬管,内填适量经处理的玻璃棉,以防止残留物进入毛细柱,并要勤换衬管为妥。 (3)因各单体对内标物的相对响应值不同,的以在配制标准溶液时,称取的各单体质量不一定都是0.03g,原则是尽量使各单体的峰面积和内标物的峰面积之比值接近于1。 (4)某些样品经丙酮稀释旋转一段时间后会出现破乳分层,做样品时须取其上层清液进色谱仪分析。 (5)醋梭乙烯酯VAM存在水的现象。因此在做其回收率时溶剂用脱水分析砘丙酮这样能有效控制VAM水解。但测试样时,溶剂为丙酮水溶液(4 1),这是由于有些样品溶于丙酮(脱水)时会产生胶化现象。
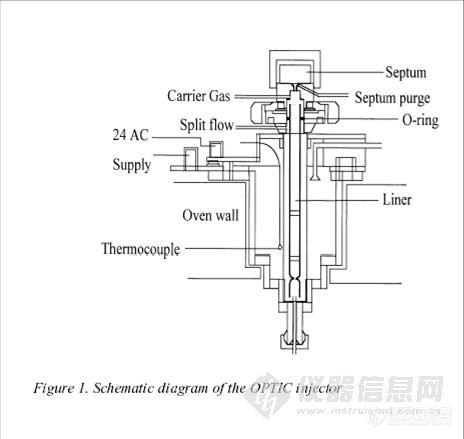
大体积进样技术用于[url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]的原理、仪器和应用 By Hans-Gerd Janssen, Hans Mol and Carel A. Cramers, Laboratory of Instrumental Analysis, Eindhoven University of Technology, P.O. Box 513, 5600 MB Eindhoven, The Netherlands.1.前言分析工作中常常需要对复杂样品中的痕量组分进行检测,这就需要在分析前对样品进行预处理。富集样品是最常采用的预处理方法,它能够满足分析方法对于检测限的要求。大体积进样技术在毛细管[url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]中的应用可以降低分析方法的检测限,提高分析灵敏度和精度。与传统的富集技术相比,大体积进样技术缩短了样品预处理时间,降低了分析费用。大体积进样和程序升温进样技术(PTV)的连用使分析操作更加快捷,分析结果更加可靠。图1为OPTIC的PTV装置图。PTV装置针对传统的分流/不分流进样方式进行了改进,增加了进样器的温控系统;PTV进样器的蒸发室可以快速加热和冷却;在低温下导入样品能够避免分流和不分流进样模式中的许多问题,使样品在进样时完全没有损失。举例来说,对于多组分样品,在低温下将样品以液体形式注入冷却后的衬管中,然后加热衬管至正常的进样温度。 [img]http://ng1.17img.cn/bbsfiles/images/2017/01/201701191651_626046_1617240_3.jpg[/img]2. PTV装置的大体积进样PTV装置操作简单,是便捷的毛细管色谱样品导入装置。PTV装置可以将高温、低温,分流和不分流进样方式完美结合,而且PTV进样装置可以选择性的排除样品中的溶剂,实现样品浓缩。在导入大体积的样品时需要控制样品的导入速度,使样品在通过分流管时实现待测组分的捕集和浓缩。在样品完全进入后,关闭分流管后,对它进行加热。这样的进样方式可以在内径为320μm的毛细管柱中导入1ml的样品。通常使用的进样速度为100-250μl/min。图2是250μl 湖水样品正己烷萃取液PTV大体积进样的色谱图。100ml的水样经2ml正己烷萃取后,取250μl的萃取液以80μl/min的速度进样。样品的检测限低于ppt级。PTV的大体积进样与[url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]联用能够将分析方法的检测限降低至原来方法的1/100。 [img]http://ng1.17img.cn/bbsfiles/images/2017/10/2009121110339_01_1617240_3.jpg[/img]图3和4列出了PTV大体积进样的另两种应用。图3为水中低含量PAH的色谱图。水样用正乙烷萃取后,以80μl/min的速度进样250μl。初始温度设置为0℃。芴、菲、芘的回收率为100%,β-甲基萘由于PTV衬管的不完全捕集,回收率为78%。 [img]http://ng1.17img.cn/bbsfiles/images/2017/10/2009121110355_01_1617240_3.jpg[/img]图4是挥发性有机物PTV大体积进样的色谱图。用Tenax填料的衬管用于捕集挥发性有机物,实现对样品中各种组分的定量回收。 [img]http://ng1.17img.cn/bbsfiles/images/2017/10/200912111049_01_1617240_3.jpg[/img]3. 结论 程序升温进样器是灵活的[url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]样品导入系统。PTV进样器是实现大体积进样的多功能型进样装置。进样速度为250μl/min时,样品的导入量可以达到1000μl。大体积进样方式的采用可以降低分析方法的检测限。OPTIC的PTV进样装置的全自动化操作可以提供可靠的分析结果,节省分析时间。
[align=center][b][size=18px]运用气相色谱内标法测定食品中甜蜜素含量 [/size][/b] [/align] 甜蜜素化学名称为环己基氨基磺酸钠,于1937年发现,1950年开始生产应用。它是由环己胺和氨基磺酸或三氧化硫反应后用NaOH处理,再重结晶制得的一种白色结晶粉末,甜度为蔗糖的30~80倍。风味较自然,后苦不明显,热稳定性高,是不被人体吸收的低热能甜味剂。1969年曾因其致畸性的报道而被世界各国禁用,后来由于大量试验表明它并无致畸、致癌等作用,许多国家重又许可使用。我国于1987年开始应用甜蜜素,它是目前我国食品行业中应用蕞多的一种甜味剂。甜蜜素含量检测目前有气相色谱检测方法、液相色谱检测方法和液相色谱-质谱/质谱法等,但应用最为广泛的方法是依据GB/T5009.97-2016《食品中环己基氨基磺酸钠的测定》标准中的气相色谱仪分析法。色谱分析技术人员采用在提取溶剂正己烷中加入两种内标物质(甲苯和乙酸正丁酯)对甜蜜素经过衍生后的产物进行定量,具有快捷,准确等特点,受到普遍欢迎。1.试验部分1.1原理在硫酸介质中甜蜜素与亚硝酸钠反应,生成环己醇亚硝酸酯,利用气相色谱法进行定性和定量。1.2试剂(所用试剂不做说明皆为分析纯,水为蒸馏水)1.2.1甜蜜素储备溶液:称取1.0000g甜蜜素(含量≥99.0%),加水溶解并定容至100mL,此溶液浓度为10.00mg/mL,为储备液。置于4℃的冰箱中。本次试验溶液浓度为:10.320mg/mL1.2.2甜蜜素标准使用溶液:取1.2.1储备液10mL,加水定容至100mL,为使用液,浓度为1.0000mg/mL。本次试验使用溶液浓度为:1.0320mg/mL1.2.3100g/L硫酸溶液:称取50g浓硫酸,用水定容至500mL。1.2.450g/L亚硝酸钠溶液:称取25g亚硝酸钠,用水定容至500mL。1.2.5正己烷1.2.6甲苯(内标)1.2.7乙酸丁酯(内标)1.2.8氯化钠1.3仪器1.3.1气相色谱仪GC-2020(带FID检测器、毛细管进样口、N2000色谱工作站)1.3.2DB-5毛细管柱(30m×0.32mm×0.25μm)或其它类型毛细管柱,如DB-1、DB-17011.3.3离心机1.3.410μL微量进样器1.4仪器操作条件1.4.1检测器:200℃1.4.2汽化室:180℃1.4.3柱温:70℃1.4.4载气流速(压力):100kPa1.4.5氢气流速(压力):50kPa1.4.6空气流速(压力):60kPa1.5样品前处理1.5.1内标溶液的配置:在正己烷中加入一定量的内标(乙酸丁酯和甲苯)1,加入的量以出色谱峰合适为宜。一般为500mL正己烷中加入内标100μL~200μL。本次实验乙酸丁酯的浓度为0.1344mg/mL(称量0.0672g乙酸丁酯于装有10mL左右正己烷的25mL容量瓶中,再将溶液转移至500mL容量瓶中并定容至刻度线,摇匀,备用。1.5.2标准溶液前处理:吸取1.2.2标准溶液10mL于50mL比色管中,加10mL水,摇匀,置于冰浴中。加入5mL50g/L亚硝酸钠溶液,5mL100g/L硫酸溶液,在冰浴中放置30min,并时常摇动,然后准确加入10mL1.5.1溶剂,5g氯化钠,摇匀后振摇80次。静置分层。吸出正己烷层于10mL带塞离心管中进行离心分层(如有机溶剂和水相很快就有分层或只要进样针能吸出提取的溶剂,就可以不需离心分离这一步骤),吸取正己烷层1μL注入色谱仪进行分析。算出校正因子2。1.5.3液体样品前处理:称取试样20.0g于50mL带塞比色管中,加10mL水。摇匀,置于冰浴中。处理过程同标准溶液前处理1.5.2。吸取正己烷层试样1μL注入色谱仪进行分析。1.5.4固体样品前处理:称取已磨碎(剪碎)试样2.0~10.0g(根据样品中甜蜜素含量而定称取质量,使甜蜜素的量在1~10mg之间)于50mL带塞比色管中,加10mL水。一些样品不易溶解,如蜜饯类、山楂等,置于水浴锅中煮沸15min左右,冷却至60℃以下,置于冰浴中。处理过程同标准溶液前处理1.5.2。吸取正己烷层试样1μL注入色谱仪进行分析。2结果与讨论2.1.1分析方法的线性相关性的测定分别吸取1.2.1标准溶液1mL、3mL、5mL、10mL、20mL于5个100mL容量瓶中,并定容至刻度,配成浓度分别为0.1032mg/mL、0.3096mg/mL、0.5160mg/mL、1.0320mg/mL、2.0640mg/mL的标准溶液。分别吸取以上标准溶液10mL于5支50mL比色管中,按1.5.2方法进行处理。每个标准点分别进样5次,每次进样1μL,以甜蜜素与内标物的质量比为横坐标,甜蜜素与内标物的峰面积比(取五次进样平均值)为纵坐标绘制标准曲线,得线性方程为y=0.43991x-0.00088,其线性相关系数为0.99999。2.1.2分析方法的准确度的测定从市场上购买一种不含甜蜜素的饮料(样品名称:鲜橙多橙汁饮料生产单位:昆山统一企业食品有限公司净含量:2L生产日期:20060307),称取5份一定量的试样,用移液管分别加入1.2.2标准溶液1、3、5、10、15mL,按上述气相色谱操作条件测定甜蜜素回收率,结果都在99.7%~101.1%之间。2.1.3分析方法的精密度的测定从市场上购买一种含有甜蜜素的饮料(样品名称:鲜橙多生产单位:康裕食品有限公司净含量:1.5L生产日期:20060415),从同一产品中称取五个试样。按1.5.2方法进行处理后,按上述气相色谱操作条件测定甜蜜素含量,得到标准偏差为0.0027,变异系数(RSD)为0.55%。3结论综上所述,本方法简便、快速、准确,具有较高的准确度、精密度和回收率,线性关系好,是一种非常可行的分析方法。注:1用甲苯和乙酸丁酯做内标都可以,可同时加入两种内标(选其中一种作为参照计算),也可以只加其中一种。本次试验的数据是以乙酸丁酯为内标进行计算而获得的。2甜蜜素用本方法衍生处理后,在毛细管柱上会出两个峰,一般是刚开始前面的大,后面的很小,但时间长了,后面的峰越来越大,相应的,前面的峰越来越小直至消失,但两个峰面积和是不变的(48小时之内)。计算校正因子时,用甜蜜素衍生产物的两个峰的面积和计算。检测样品时,两个峰可单独识别并采用同一个校正因子计算,结果相加,也可以使用工作站将峰面积相加,再计算含量。一批样品检测,样品和标准品使用同一浓度的内标溶剂,在计算校正因子时,可将内标物的量假定为1(任一定值皆可),使用非常方便,不需知道加入的内标物的质量。否则,要称量内标物的质量,[font=FZSSK--GBK1-0]精确[/font]至0.0001g,再定容至一定体积。算出内标物浓度,再进行校正因子的计算。
[url=https://insevent.instrument.com.cn/t/Mp]气相色谱仪[/url]测量呼吸量时,如何测量不规则样品的体积?
用[url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]如何测定气体中的水分含量?样品:氯化氢与乙炔的混合气;水分含量:约0.06%左右;若哪位大哥知道,请告诉我色谱柱型号,填充料,温控数据,其他操作条件。谢谢了!
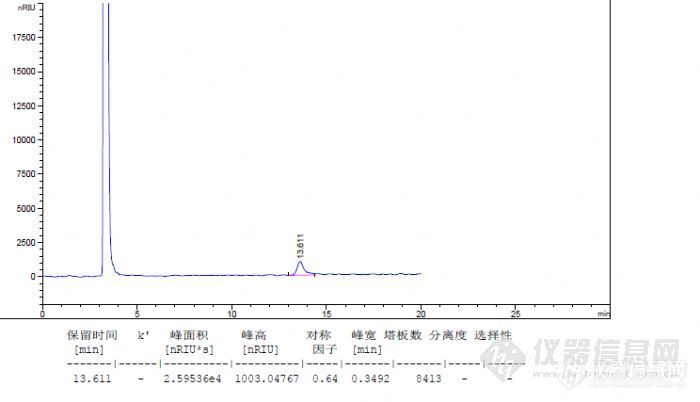
前言:按照10版药典方法检测乳糖的含量,按照系统适应性下的要求,乳糖与蔗糖达到分离要求,乳糖的理论踏板数不小于5000。下面主要考察乳糖在测试过程中,相同样品浓度下不同的进样体积对柱效的影响和不同浓度下相同进样体积对柱效的影响。测定方法:色谱柱:Ultimate®XB-NH2-3,4.6×250mm,5μm; 温度:柱温:45度,示差检测器:40度;流动相:乙腈-水=70:30; 流速:1.0mL/min; 进样量:10μL。样品配制:系统适用性:精密称取乳糖0.0255g和蔗糖0.0254g置于25ml容量瓶中加水溶解并稀释至刻度摇匀,过滤,即得。含量测定样品溶液:精密称取样品粉末0.0252g于25ml容量瓶中,用水溶解并定容,过滤即得;含量测定对照品溶液:精密称取对照品粉末0.0254g于25ml容量瓶中,用水定容,过滤即得按照测定要求,按照测定要求样品浓度为1mg/ml,进样体积:10ul考虑到我的仪器的定量环是20ul,因为是手动进样,进样量10ul可能在计算含量的时候峰面积不稳定计算会产生误差。考虑把样品浓度配制成0.5mg/ml,调整进样体积为20ul;下面上一张系统适用性图:http://ng1.17img.cn/bbsfiles/images/2012/11/201211032333_401206_1938106_3.jpg重复了好几针,分离度和保留时间没改变,柱效依然达不到5000.乳糖对照品0.5mg/ml 进样量20ulhttp://ng1.17img.cn/bbsfiles/images/2012/11/201211032338_401211_1938106_3.jpg测试乳糖对照品,乳糖的柱效也是比较低的。考虑到示差检测器是比较难稳定的,也考虑氨基柱在平衡中是比较难平衡的,所以采用过夜循环平衡方法进行测定。测试乳糖对照品0.5mg/ml 进样量20ulhttp://ng1.17img.cn/bbsfiles/images/2012/11/201211040004_401226_1938106_3.jpg平衡了一夜但的柱效一点都没有改善。柱效依然不能满足要求。这个过程考虑到是否是氨基柱的问题,排除了氨基柱在使用过程中的氨基脱落导致柱效的原因,为了排除色谱柱的原因换新柱子测试,柱效还是不能满足要求。百思不得其解中,想到样品质量对柱效影响,还是进样体积对柱效是否有影响。为了证明是相同质量下,不同进量是否对柱效存在影响。下面配制1mg/ml的浓度分别进样20ul和10ul的体积。乳糖对照品1.0mg/ml 进样量20ul http://ng1.17img.cn/bbsfiles/images/2012/11/201211041842_401303_1938106_3.jpg乳糖对照品1.0mg/ml 进样量10ulhttp://ng1.17img.cn/bbsfiles/images/2012/11/201211041848_401305_1938106_3.jpg进样体积减少10ul发现柱效提高一倍多。结论:从以上实验结果分析比较当乳糖含量样品体积为20ul,样品浓度1mg/ml,柱效:3982;0.5mg/ml,浓度,柱效4253;当乳糖浓度1mg/ml的浓度进样为20ul柱效:3982;进样量10ul柱效:8413,进样体积减小一半,柱效提高一倍。说明在夜想色谱柱具有一定的饱和容量下,最大进样体积,也要决定于化合物在固定相和流动相中平衡,通过上面实验证明,随着进样量的增大,柱效降低。当进样量超出色谱柱饱和容量时,柱效也会降低。对于一定的样品量,进样体积增大,柱效降低。当进样量超出色谱柱上样最大进样体积时,柱效会有明显降低,影响分离结果。当然对于分析色谱而言,所需要的进样量和进样体积都是很小的,所以在平时很少观察到这点现象。可以看出进样量和进样体积对定性定量中对色谱柱柱效的影响。发现晚上传图太慢了, 所以今天晚上才把图传上来,供各位色友分享。
李石钿肇庆市建设工程质量检测站广东肇庆 526020摘要:随着人们生活水平的不断改善,环境意识的增强,人们对室内环境的空气质量日益关注。而室内环境空气中的TVOC含量将直接影响着室内环境的空气质量。本文结合测定实例,阐述了气相色谱法在测定室内环境空气TVOC中的应用,介绍了气相色谱法的操作步骤,取得了准确的测定结果。关键词:TVOC;气相色谱法;操作步骤;结果分析 随着人们生活水平的提高,人们对居住环境的装饰效果也要求更高,各种装饰装修材料进入室内,由此使室内空气污染物的来源和种类越来越多。特别是室内装修中,使用的劣质材料、劣质油漆黏合剂、加上劣质家具,使我们的室内空气中挥发性有机毒物增加,严重影响我们的身心健康和生命安全。由于TVOC 在空气中的含量很低,在进行GC 测定之前,须对其进行吸收浓缩,而后在选定条件下热解吸,并对热解吸所产生的气体进行GC 或GC-MS 测定。其中,热解吸时间,温度和解吸气体进样的时间是三项影响测定的重要因素。通过实验对上述重要因素确定了合理的控制值。结果证实可以取得稳定而满意的测定数据。 1 实验条件 仪器设备: HITACHI G-3900 气相色谱仪; 太极计算机公司TJ-618 热解吸仪; SE30 色谱柱/50 m/0.32 mm/1 μm;氢火焰离子化检测器。 标准品: TVOC 标液,上海师范大学。色谱柱升温程序: 初温50 ℃、保持5 min,5 ℃/min、升至250 ℃、保持2 min。热解吸升温程序: 初温40 ℃,分别升至150 ℃ /200 ℃ /250 ℃ /300 ℃,经热解吸各3 min /4 min /5 min /6 min 后开始进样,直至分析测定结束。 2 实验结果与讨论 2.1 热解吸进样时间对被测组分峰面积的影响 TVOC 标液被Tenax 吸收管吸附后,置入热解吸器,使其迅速升温至250 ℃。接着分别保持3 min,4 min,5 min,6 min 进行热解吸。将解吸产生的TVOC 组分导入气相色谱仪,所得各组分的峰面积与热解吸时间的关系,分别见图1,图2。[/fon
[em0716] 现在有的条件是 6890 [url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url],载气为氮气,多种组分的气体为:氢气、甲烷、氮气、氧气、一氧化碳、二氧化碳。请教问题: 1、气体如何收集? 待检测的气体流量不是很大。现有的收集设备是10ml Aglient的进样针,不知取样后待测气体是否会被空气所污染? 2、色谱需要设定怎样的条件? 做了几个样,由于用氮气作为载气,检测出的氢气峰是倒峰,而且出现的峰主要只有一个,估计是氢气,几个样的峰尖位置时间都不在同一点,不知道什么原因造成? 3、如何通过出现的峰来反应出氢气的含量? 从实验条件考虑,用外表法得到特征曲线(具体用空气稀释纯氢得到不同含量的氢气标样),此方法是否合适?是否适用此种多组分的气体?或者是否有更好的方法? 谢谢帮忙解决@!!
现车间排放的尾气中含有少量的NMP(N-甲基吡咯烷酮),大概几百PPM,其他都是空气。现领导要求用气相色谱法测试尾气中NMP的含量。现在的难点在于NMP在常温下是高沸点的液体(沸点有200多度),所以用内标法时找不到合适的内标物及气体NMP标样。现在我想到一个方法不知可行不:1.用注射器取定量体积的尾气注射于专门的气体收集袋中;2.再用注射器将与NMP互溶的溶剂(比如丙酮)适量注射于气体收集袋中,充分摇匀,目的是使尾气中的NMP全部溶解于丙酮中;3.再取出溶解有NMP的丙酮,运用常规内标法测试NMP含量。 不知以上方法可行不?我最担心的是丙酮是否能将气体袋中的NMP全部溶解下来?如果不行,那这个方法就是行不通的,请教高手指点,或者用气相色谱法分析此类样品有什么其他更好的办法。谢谢!
[color=#444444]求问怎样采用[url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]测量样品气体中的甲醇含量,[/color][color=#444444]比如标样怎么配?配液体标样可以吗?[/color][color=#444444]液体标样怎样配?标样浓度单位和我的气体样品浓度单位怎样统一?[/color][color=#444444][url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]参数怎样设置?[/color]
SC-II型[url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]测定的气体厘米长度,怎么换算成气体含量啊?急!!拜托了!!
请教各位大佬。为什么我们用面积归一方法打出来的含量和实际不一样。我们用的是安捷伦7820A [table=720][tr][td=10,1]色谱柱:Agilent 19091Z-215:325℃: 50m×320μm×1.05μm 流速:3 mL/min 压力21.706psi[/td][/tr][tr][td=10,1]进样:后SSZ进样口N2[/td][/tr][tr][td=10,1]出样:后检测器FID 火焰:开[/td][/tr][tr][td=10,1]检测器加热器:300℃ H2流量:30 mL/min 空气流量:400mL/min 尾吹N2流量:25mL/min [/td][/tr][tr][td=10,1]自动进样:进样针规格10μL,进样量0.4μL×1[/td][/tr][tr][td=10,1]溶剂A清洗:进样前2次进样后5次 体积最大[/td][/tr][tr][td=10,1]样品清洗次数:4次,体积最大,样品抽吸次数5次[/td][/tr][tr][td=10,1]进样口分流,加热器温度280℃,压力21.706psi 分流比30:1 流量90mL/min [/td][/tr][tr][td=10,1]载气节省:关闭,流量20ml/min 开始等待时间2分钟[/td][/tr][tr][td=10,1]柱箱温度:开,最高柱箱温度425℃,初始温度100℃升温速率10℃/min最终温度260℃ 时间3min[/td][/tr][/table]我们打的产品是TMDD+乙二醇的复配产品, 配比基本上是在1:1,但是出峰的数据 [table=249][tr][td]3.028[/td][td]25.16 [/td][td]12.048[/td][td]74.62[/td][/tr][/table]在相应的出峰时间上乙二醇是25.16% TMDD是74.62%。按照常规理解应该是一半一半的含量。色谱小白不知道是什么原因,造成这样的结果,打样的时候产品有点稠有用酒精稀释,×掉酒精峰后是上面的结果,有尝试过不用酒精稀释直接进样打平行样看是否能够避免实际含量不一致的情况,但是出来的结果除了第一次第二次接近实际的配比,到第三次后数据就发生了很大的变化,感觉直接进样也是不行。现在不知道该怎么办,想知道还有什么办法能够准确定量产品的含量?也想知道为什么会出现上面含量和实际不一致的情况,我想知道大家有没有碰到过,都是怎么解决的。小白在这先谢谢大家指导!
我公司想购买[url=https://insevent.instrument.com.cn/t/Mp]气相色谱仪[/url]用于抽检采购产品的质量,主要是检测溶剂的确实含量,不知那个国产或进口的牌子、技术支援好点?还有,做这样的检测工作,是用标准品来标定吗?单单用峰面积计算行不行呢?
[color=#444444]如题,一般做[url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]都是有机气体和无机气体的谱图分别显示,那么如果我想将有机气体和无机气体的百分含量都折算到一起,即两者总和加起来是100%需要如何处理啊?请高手指点一二!![/color]
专家你好,我用的是ov101的毛细管柱,能否分开正己烷和乙酸乙酯,条件是什么?气谱打出的是质量含量,我要想知道体积比,还需要换算是吗?
合成树脂乳液中残余单体含量的气相色谱测定方法探讨 吴亚虎,韩婷婷(广东中山市巴德士化工有限公司品控中心,528427) 摘要:讨论了用气相色谱内标法测定合成树脂乳液中未反应完的残余单体含量的方法,并对该方法的精密度、准确度进行了考察/实验表明,该方法简单易行,准确可靠,适用于各种合成树脂乳液样品。 关键词:气相色谱;极性小口径毛细管柱;内标法; 醋酸乙烯酯、甲基丙烯酸甲酯、苯乙烯、丙烯酸丁酯、丙烯酸异辛酯等单体。 0.引言 自国家十项强制性标准颁布以后,市面上各种涂料中的有害物质必须达到国家相关限量标准。由于合成树脂乳液中未反应的残余单体对人的身体健康和环境会带来不同程度的影响,为此必须设法控制乳液中残余单体的浓度……..目前国科多采用顶空进样技术分析乳液中的残余单体含量,由于仪器投资较大,且样品回收率也不理想,样品前处理较烦锁,对于中小企业这样投资较少,易于在中小企业中推广,该分析方法较难于推广,而我们介绍的是普通分析方法。采用小口径毛细管柱,用氢火焰离子化检测器(FID)进行检测,以内标法定量。柱温采用程序升温,分离效果十分理想。其加标回收率分别在94%-103%之间,分析结果的精密度、准确度完全达到检测要求。 1.实验部分: 1.1 仪器和试剂 电子分析天平(万分之一);GC5890F气相色谱仪(带有分流装置);1.0ul微量进样器;20ml带胶塞小玻璃瓶若干;医用注射器1ml、2ml各两只;小口径毛细柱DB-17HT(0.25mm x 30m x 0.15um;最高使用温度为360℃);积分仪或色谱工作站。醋酸乙烯酯VAM(色谱纯)、甲基丙烯酸甲酯MMA(色谱纯)、苯乙烯ST(色谱纯)、丙烯酸丁酯BA(色谱纯)丙烯异辛酯2-EHA(色谱纯)、丙酮(分析纯)配成4+1混合水溶液(作稀释剂),水(纯净水或蒸馏水)。内标物:环已酮(色谱纯) 1.2 测定原理试样中加适量内标物并用少许丙酮(4+1)稀释摇匀后,用微量注射器将稀释后的溶液注入气相色谱仪,样品被载气带入色谱柱,在柱内被分离成相应的组份,用氢火焰离子化检测器检测并记录色谱图,反数据用内标法计算试样溶液中各待测残余单体的含量。 1.3 测定条件 气化温度:280℃ 检测温度:320℃ 载气:氮气:纯度≥99.99%,变色硅胶+5A分子筛除水、除油,柱前压力为60Kpa(30℃); 氢气:纯度≥99.99%,变色硅胶+5A分子筛除水、除油,柱前压力为65Kpa(30℃); 空气:变色硅胶[/fon
大家好,我在用高效液相色谱测芒果苷含量,具体步骤是:取干燥叶片0.1g,加25ML甲醇(萃取剂),称重,超声提取后冷却,用甲醇补足失质量。旋转蒸发仪加热回流,蒸干物用甲醇溶解(这一步是补足失掉甲醇量还是定容到25ML?),最后过滤,滤液体积v1,统一取滤液10ML进行液相色谱测。我只知道液相色谱可以测出芒果苷浓度,但是回归到叶片怎么计算芒果苷含量?哪些体积量是需要用到的。研一新手,实在是搞不懂,恳请大家帮助,谢谢!
[url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]绘制曲线时发现峰面积和含量差不多一样的,如苯的含量为1它的峰面积就大概为0.9~1.5左右。这是什么问题?求助[img]https://ng1.17img.cn/bbsfiles/images/2020/03/202003132239344942_5520_3869857_3.png[/img]
我用的是TCD监测器,一个[url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]柱能同时测出O2、N2、CH4、CO;所测气体中应该无氧气,若有就是混进的空气,计算时想把空气从中去除,该怎么算?若被测气体中有的成分测不出,所能测气体的所测百分含量与实际百分含量比会怎样变化?
我们原来是用电位滴定法测糠醛含量的,现在我指导老师让我用气相色谱测,具体像用什么柱子,温度怎么设置,不太清楚,求大家给点建议哈~~~~
请问一般来说0.06%高氯酸是百分含量还是体积含量?我们买的分析纯的高氯酸含量是70-72%,是不是还要进行换算?
你好,我想咨询一下有关硝基苯含量的测定方法问题:如果用极谱法测定苯胺中微量硝基苯含量的具体条件是什么??如果用[url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]法呢???







 我要推广仪器
我要推广仪器
 下载APP
下载APP