大家好,我在用高效液相色谱测芒果苷含量,具体步骤是:取干燥叶片0.1g,加25ML甲醇(萃取剂),称重,超声提取后冷却,用甲醇补足失质量。旋转蒸发仪加热回流,蒸干物用甲醇溶解(这一步是补足失掉甲醇量还是定容到25ML?),最后过滤,滤液体积v1,统一取滤液10ML进行液相色谱测。我只知道液相色谱可以测出芒果苷浓度,但是回归到叶片怎么计算芒果苷含量?哪些体积量是需要用到的。研一新手,实在是搞不懂,恳请大家帮助,谢谢!
[color=#444444]液相色谱,在不进对照品情况下,如何根据液相图谱面积百分比计算出目标峰的含量?[/color]
液相色谱,有结晶水的原料药怎样计算含量我们液相色谱测样品的含量,样品中含两个结晶水,但所用对照品是无水的,计算样品含量时要扣除那两分子的水分吗?
高效液相色谱法测定黄芪中黄芪甲苷含量的计算方法
有内标物时高效液相色谱法中如何计算含量
各位老师!用高效液相色谱仪检测纺织品邻苯二甲酸酯如何计算其含量?
求助~液相色谱含量检测的具体计算方法知道的说一下,谢谢!
各位师傅!用高效液相色谱仪检测纺织品邻苯二甲酸酯如何计算含量。
如题,液相色谱中,如何判定计算测的样品中含某一邻苯二甲酸盐的含量?比如,我这个样品测的图谱与标准品对比,并且确定这个峰就是DEHP,那么我应该怎么计算出这个样品中含DEHP的含量是多少?
用液相色谱分析废水中酚的含量,进样时酚标液应配成多大浓度?测废水中酚含量时,是直接用废水中酚与酚标液的峰峰面积比计算呢,还是将酚标液配成不同浓度作出标准曲线,从曲线上读呢?谢谢
[url=https://insevent.instrument.com.cn/t/5p][color=#3333ff]液相色谱[/color][/url]含量测定中测的值为98%,实际含量为96%,一个称量0.1g,稀释100倍,一个称量0.01g稀释10倍,结果前者测得含量98,后者测得含量97,这个是什么原因
高效液相色谱分析血液中儿茶酚胺含量,回收率不高各位老师,我想要用用高效液相色谱分析血液中儿茶酚胺含量,用的是电化学检测器,柱子是C18柱,用氧化铝吸附解析,找了几篇论文但是回收率不高,不知道是柱子没选对,还是前处理做的不好,或者是流动相不好,各位老师有什么意见可以知道一下吗?
[b]高效液相色谱法的计算方法[/b]高效液相色谱法是用高压输液泵将具有不同极性的单一溶剂或不同比例的混合溶剂、缓冲液等流动相泵入装有固定相的色谱柱,经进样阀注入供试品,由流动相带入柱内,在柱内各成分被分离后,依次进入检测器,色谱信号由记录仪或积分仪记录。 1.对仪器的一般要求 所用的仪器为高效液相色谱仪。色谱柱的填料和流动相的组分应按各品种项下的规定.常用的色谱柱填料有硅胶和化学键合硅胶。后者以十八烷基硅烷键合硅胶最为常用,辛基键合硅胶次之,氰基或氨基键合硅胶也有使用;离子交换填料,用于离子交换色谱;凝胶或玻璃微球等,用于分子排阻色谱等。注样量一般为数微升。除另有规定外,柱温为室温,检测器为紫外吸收检测器。 在用紫外吸收检测器时,所用流动相应符合紫外分光光度法(附录Ⅳ A)项下对溶剂的要求。 正文中各品种项下规定的条件除固定相种类、流动相组分、检测器类型不得任意改变外,其余如色谱柱内径、长度、固定相牌号、载体粒度、流动相流速、混合流动相各组分的比例、柱温、进样量、检测器的灵敏度等,均可适当改变, 以适应具体品种并达到系统适用性试验的要求。一般色谱图约于20分钟内记录完毕。 2.系统适用性试验 按各品种项下要求对仪器进行适用性试验,即用规定的对照品对仪器进行试验和调整,应达到规定的要求;或规定分析状态下色谱柱的最小理论板数、分离度和拖尾因子. (1) 色谱柱的理论板数(n) 在选定的条件下,注入供试品溶液或各品种项下规定的内标物质溶液,记录色谱图,量出供试品主成分或内标物质峰的保留时间t(以分钟或长度计,下同,但应取相同单位)和半高峰宽(W),按n=5.54(t/W)计算色谱柱的理论板数,如果测得理论板数低于各品种项下规定的最小理论板数,应改变色谱柱的某些条件(如柱长、载体性能、色谱柱充填的优劣等),使理论板数达到要求。 (2) 分离度 定量分析时,为便于准确测量,要求定量峰与其他峰或内标峰之间有较好的分离度。分离度(R)的计算公式为: 2(t-t) R= ──W+W 式中 t为相邻两峰中后一峰的保留时间; t为相邻两峰中前一峰的保留时间; W及W为此相邻两峰的峰宽。 除另外有规定外,分离度应大于1.5。 (3) 拖尾因子 为保证测量精度,特别当采用峰高法测量时,应检查待测峰的拖尾因子(T)是否符合各品种项下的规定,或不同浓度进样的校正因子误差是否符合要求。拖尾因子计算公式为: W T=────── 2d 式中 W为0.05峰高处的峰宽; d为峰极大至峰前沿之间的距离。 除另有规定外,T应在0.95~1.05间。 也可按各品种校正因子测定项下,配制相当于80%、100%和120%的对照品溶液,加入规定量的内标溶液,配成三种不同浓度的溶液,分别注样3次,计算平均校正因子,其相对标准偏差应不大于2.0%。 3.测定法 定量测定时,可根据样品的具体情况采用峰面积法或峰高法。但用归一法或内标法测定杂质总量时,须采用峰面积法。 (1) 面积归一化法 测定供试品(或经衍生化处理的供试品)中各杂质及杂质的总量限度采用不加校正因子的峰面积归一法。计算各杂质峰面积及其总和,并求出占总峰面积的百分率。但溶剂峰不计算在内。色谱图的记录时间应根据各品种所含杂质的保留时间决定,除另有规定外,可为该品种项下主成分保留时间的倍数。 (2) 主成分自身对照法 当杂质峰面积与成分峰面积相差悬殊时,采用主成分自身对照法。在测定前,先按各品种项下规定的杂质限度,将供试品稀释成一定浓度的溶液作为对照溶液,进样,调节检测器的灵敏度或进样量,使对照溶液中的主成分色谱峰面积满足准确测量要求。然后取供试品溶液,进样,记录时间,除另有规定外,应为主成分保留时间的倍数。根据测得的供试品溶液的各杂质峰面积及其总和并和对照溶液主成分的峰面积比较,计算杂质限度。 (3) 内标法测定供试品中杂质的总量限度 采用不加校正因子的峰面积法。取供试品,按各品种项下规定的方法配制不含内标物质的供试品溶液,注入仪器,记录色谱图I 再配制含有内标物质的供试品溶液,在同样的条件下注样,记录色谱图Ⅱ。记录的时间除另有规定外,应为该品种项下规定的内标峰保留时间的倍数,色谱图上内标峰高应为记录仪满标度的30%以上,否则应调整注样量或检测器灵敏度。 如果色谱图Ⅰ中没有与色谱图Ⅱ上内标峰保留时间相同的杂质峰,则色谱图Ⅱ中各杂质峰面积之和应小于内标物质峰面积(溶剂峰不计在内)。如果色谱图Ⅰ中有与色谱图Ⅱ上内标物质峰保留时间相同的杂质峰,应将色谱图Ⅱ上的内标物质峰面积减去色谱图Ⅰ中此杂质峰面积,即为内标物质峰的校正面积;色谱图Ⅱ中各杂质峰总面积加色谱图Ⅰ中此杂峰面积,即为各杂质峰的校正总面积,各杂质峰的校正总面积应小于内标物质峰的校正面积。 (4) 内标法加校正因子测定供试品中某个杂质或主成分含量 按各品种项下的规定,精密称(量)取对照品和内标物质,分别配成溶液,精密量取各溶液,配成校正因子测定用的对照溶液,取一定量注入仪器,记录色谱图,测量对照品和内标物质的峰面积或峰高,按下式计算校正因子: A/m 校正因子(f)=─ A/m 式中 A为内标物质的峰面积或峰高;A为对照品的峰面积或峰高; m为加入内标物质的量; m为加入对照品的量。 再取各品种项下含有内标物质的供试品溶液,注入仪器,记录色谱图,测量供试品(或其杂质)峰和内标物质的峰面积或峰高,按下式计算含量:A 含量(m)=f×──A/m 式中 A为供试品(或其杂质)峰面积或峰高;m为供试品(或其杂质)的量; f、A和m的意义同上。 当配制校正因子测定用的对照溶液和含有内标物质的供试品溶液使用同一份内标物质溶液时,则配制内标物质溶液不必精密称(量)取。 (5) 外标法测定供试品中某个杂质或主成分含量,按各品种项下的规定,精密称(量)取对照品和供试品,配制成溶液,分别精密取一定量,注入仪器,记录色谱图,测量对照品和供试品待测成分的峰面积(或峰高),按下式计算含量:A 含量(m)=m×── A 式中各符号意义同上由于微量注射器不易精确控制进样量,当采用外标法测定供试品中某杂质或主成分含量时,以定量环进样为好。来源:分析化学网。[em61]
高效液相色谱法的计算方法高效液相色谱法是用高压输液泵将具有不同极性的单一溶剂或不同比例的混合溶剂、缓冲液等流动相泵入装有固定相的色谱柱,经进样阀注入供试品,由流动相带入柱内,在柱内各成分被分离后,依次进入检测器,色谱信号由记录仪或积分仪记录。1.对仪器的一般要求所用的仪器为高效液相色谱仪。色谱柱的填料和流动相的组分应按各品种项下的规定.常用的色谱柱填料有硅胶和化学键合硅胶。后者以十八烷基硅烷键合硅胶最为常用,辛基键合硅胶次之,氰基或氨基键合硅胶也有使用;离子交换填料,用于离子交换色谱;凝胶或玻璃微球等,用于分子排阻色谱等。注样量一般为数微升。除另有规定外,柱温为室温,检测器为紫外吸收检测器。在用紫外吸收检测器时,所用流动相应符合紫外分光光度法(附录Ⅳ A)项下对溶剂的要求。正文中各品种项下规定的条件除固定相种类、流动相组分、检测器类型不得任意改变外,其余如色谱柱内径、长度、固定相牌号、载体粒度、流动相流速、混合流动相各组分的比例、柱温、进样量、检测器的灵敏度等,均可适当改变,以适应具体品种并达到系统适用性试验的要求。一般色谱图约于20分钟内记录完毕。2.系统适用性试验按各品种项下要求对仪器进行适用性试验,即用规定的对照品对仪器进行试验和调整,应达到规定的要求;或规定分析状态下色谱柱的最小理论板数、分离度和拖尾因子.(1) 色谱柱的理论板数(n)在选定的条件下,注入供试品溶液或各品种项下规定的内标物质溶液,记录色谱图,量出供试品主成分或内标物质峰的保留时间t(以分钟或长度计,下同,但应取相同单位)和半高峰宽(W),按n=5.54(t/W)计算色谱柱的理论板数,如果测得理论板数低于各品种项下规定的最小理论板数,应改变色谱柱的某些条件(如柱长、载体性能、色谱柱充填的优劣等),使理论板数达到要求。(2) 分离度定量分析时,为便于准确测量,要求定量峰与其他峰或内标峰之间有较好的分离度。分离度(R)的计算公式为: 2(t-t) R= ──W+W 式中 t为相邻两峰中后一峰的保留时间; t为相邻两峰中前一峰的保留时间; W及W为此相邻两峰的峰宽。 除另外有规定外,分离度应大于1.5。(3) 拖尾因子 为保证测量精度,特别当采用峰高法测量时,应检查待测峰的拖尾因子(T)是否符合各品种项下的规定,或不同浓度进样的校正因子误差是否符合要求。拖尾因子计算公式为: W T=────── 2d 式中 W为0.05峰高处的峰宽; d为峰极大至峰前沿之间的距离。 除另有规定外,T应在0.95~1.05间。 也可按各品种校正因子测定项下,配制相当于80%、100%和120%的对照品溶液,加入规定量的内标溶液,配成三种不同浓度的溶液,分别注样3次,计算平均校正因子,其相对标准偏差应不大于2.0%。3.测定法 定量测定时,可根据样品的具体情况采用峰面积法或峰高法。但用归一法或内标法测定杂质总量时,须采用峰面积法。 (1) 面积归一化法 测定供试品(或经衍生化处理的供试品)中各杂质及杂质的总量限度采用不加校正因子的峰面积归一法。计算各杂质峰面积及其总和,并求出占总峰面积的百分率。但溶剂峰不计算在内。色谱图的记录时间应根据各品种所含杂质的保留时间决定,除另有规定外,可为该品种项下主成分保留时间的倍数。(2) 主成分自身对照法当杂质峰面积与成分峰面积相差悬殊时,采用主成分自身对照法。在测定前,先按各品种项下规定的杂质限度,将供试品稀释成一定浓度的溶液作为对照溶液,进样,调节检测器的灵敏度或进样量,使对照溶液中的主成分色谱峰面积满足准确测量要求。然后取供试品溶液,进样,记录时间,除另有规定外,应为主成分保留时间的倍数。根据测得的供试品溶液的各杂质峰面积及其总和并和对照溶液主成分的峰面积比较,计算杂质限度。(3) 内标法测定供试品中杂质的总量限度采用不加校正因子的峰面积法。取供试品,按各品种项下规定的方法配制不含内标物质的供试品溶液,注入仪器,记录色谱图I 再配制含有内标物质的供试品溶液,在同样的条件下注样,记录色谱图Ⅱ。记录的时间除另有规定外,应为该品种项下规定的内标峰保留时间的倍数,色谱图上内标峰高应为记录仪满标度的30%以上,否则应调整注样量或检测器灵敏度。如果色谱图Ⅰ中没有与色谱图Ⅱ上内标峰保留时间相同的杂质峰,则色谱图Ⅱ中各杂质峰面积之和应小于内标物质峰面积(溶剂峰不计在内)。如果色谱图Ⅰ中有与色谱图Ⅱ上内标物质峰保留时间相同的杂质峰,应将色谱图Ⅱ上的内标物质峰面积减去色谱图Ⅰ中此杂质峰面积,即为内标物质峰的校正面积;色谱图Ⅱ中各杂质峰总面积加色谱图Ⅰ中此杂峰面积,即为各杂质峰的校正总面积,各杂质峰的校正总面积应小于内标物质峰的校正面积。(4) 内标法加校正因子测定供试品中某个杂质或主成分含量按各品种项下的规定,精密称(量)取对照品和内标物质,分别配成溶液,精密量取各溶液,配成校正因子测定用的对照溶液,取一定量注入仪器,记录色谱图,测量对照品和内标物质的峰面积或峰高,按下式计算校正因子: A/m 校正因子(f)=─ A/m 式中 A为内标物质的峰面积或峰高;A为对照品的峰面积或峰高; m为加入内标物质的量; m为加入对照品的量。再取各品种项下含有内标物质的供试品溶液,注入仪器,记录色谱图,测量供试品(或其杂质)峰和内标物质的峰面积或峰高,按下式计算含量:A 含量(m)=f×──A/m 式中 A为供试品(或其杂质)峰面积或峰高;m为供试品(或其杂质)的量; f、A和m的意义同上。当配制校正因子测定用的对照溶液和含有内标物质的供试品溶液使用同一份内标物质溶液时,则配制内标物质溶液不必精密称(量)取。(5) 外标法测定供试品中某个杂质或主成分含量,按各品种项下的规定,精密称(量)取对照品和供试品,配制成溶液,分别精密取一定量,注入仪器,记录色谱图,测量对照品和供试品待测成分的峰面积(或峰高),按下式计算含量:A 含量(m)=m×── A 式中各符号意义同上由于微量注射器不易精确控制进样量,当采用外标法测定供试品中某杂质或主成分含量时,以定量环进样为好。来源:分析化学网。
有没有分析3-氨基丙腈的液相色谱分析方法我想用液相色谱分析有机盐中的3-氨基丙腈的含量,但由于3-氨基丙腈在紫外和示差液相中响应值不是很大,满足不了微量杂质的分析。大家有没有好的建议。比如有没有合适的检测器、或则有合适的衍生法。(由于样品中含有氨基丙酸,所以氨基的衍生手段好像无法使用)
[color=#444444]请问用液相色谱串联质谱法测试水样中农药含量时,为什么要在过滤后的水样中加入等体积的有机溶剂?最终如何计算测得的浓度?是否为2倍关系?[/color]
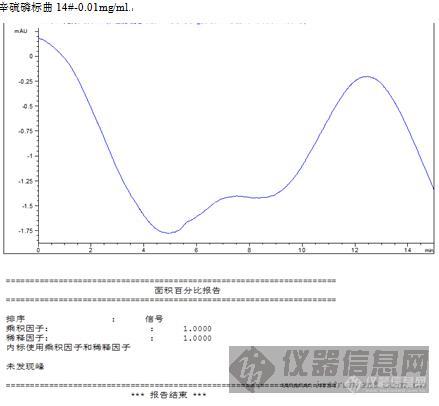
初接触液相色谱,做土壤农残含量测定得到液相色谱图不知道怎么计算出含量?请各位指教。
最近我化验室的同事在讨论使用高效液相色谱检测日常样品时是否有必要计算校正因子的RSD,如果要需要的话,那么RSD要控制在多少以下?在书上有规定吗?请问各位你们日常检测样品时是否也有计算校正因子的RSD呢?
液相色谱能否用有效碳数规律来计算质量校正因子
高效液相色谱分析废水中氯苯的含量的毕业设计怎么做?我要做毕业设计可是具体怎么做我还是不清楚,希望哪们高手能帮帮忙!
液相色谱检测克百威的含量是用到正向柱时,发现压力正负不停变化,时什么原因,液相色谱室岛津的LC-20AT,单向阀已经拆下来用异丙醇超声震荡1小时了,流动相是99.3:0.7的正己烷和四氢呋喃,求老师指导,谢谢了
[color=#444444]近来准备用高效液相色谱测葡萄糖的含量,第一次做这种实验,没有多少经验,忘大神指点啊!!!波长多少?流动相的选择?色谱柱?[/color]
如何用高效液相色谱检测松伯醇的含量多少?
请问怎么用液相色谱测定样品中的奈含量色谱仪不限 样品不限最好能附操作步骤谢谢大家!!

使用高效液相色谱测定水中甲酸含量,前面为什么会出现?做标准曲线的时候还蛮准的,但是测样品的时候基线下凹(图中基线是人为拉直了),含量测不准确?http://ng1.17img.cn/bbsfiles/images/2015/04/201504011537_540437_2966883_3.jpghttp://ng1.17img.cn/bbsfiles/images/2015/04/201504011537_540438_2966883_3.jpg
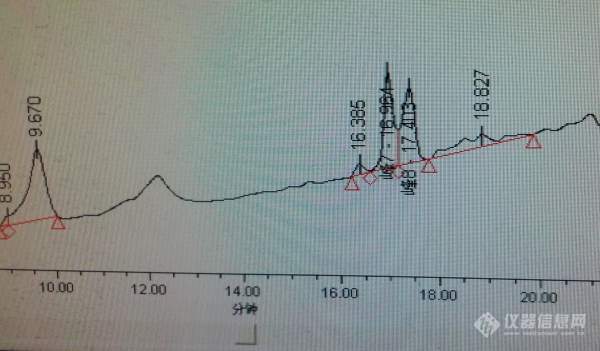
[color=#444444]在使用液相色谱测定环己烷中蒽的含量时,采用的是在文献《有机脱模剂中多环芳烃的高效液相色谱测定》中的方法,温度是20度,检测波长210 nm,采用梯度淋洗方式,开始时,流动相为 (乙腈): (水)=40:60,28 min后,变为V(乙腈):V(水)=82:18,48 min后,变成(乙腈):V(水)=100:0,保持8 min。流速为1ml/min,进样量为10微升。但是这样做走基线之后测出来的图线是飘的,基线偏移很厉害,虽然在17min附近蒽的峰会出现,但是出现的是双峰。我使用的是150mm的C18柱。[/color][color=#444444]师姐说我是因为采用了梯度淋洗的方式所以才会基线偏移,我昨天试了一下直接进V(乙腈):V(水)=70:30的流动相,虽然基线不漂了,但是峰都重叠在一起,全部在7min之前就出来了,我也不知道哪个是蒽,而且峰型都很差。[/color][color=#444444]有没有方法可以在我不改变梯度淋洗的方法的情况下,不出现基线漂移的现象?[/color][color=#444444]或者是有其他更好的流动相进样方法?[/color][color=#444444]我之前出的结果都是双峰,但我不是很想再重做了,这些数据可以用吗?我用的外标法,不知道计算是算两个双峰面积之和还是算平均?[/color][color=#444444][img=,600,351]https://ng1.17img.cn/bbsfiles/images/2019/09/201909231135324988_5836_1849104_3.jpg!w600x351.jpg[/img][/color]
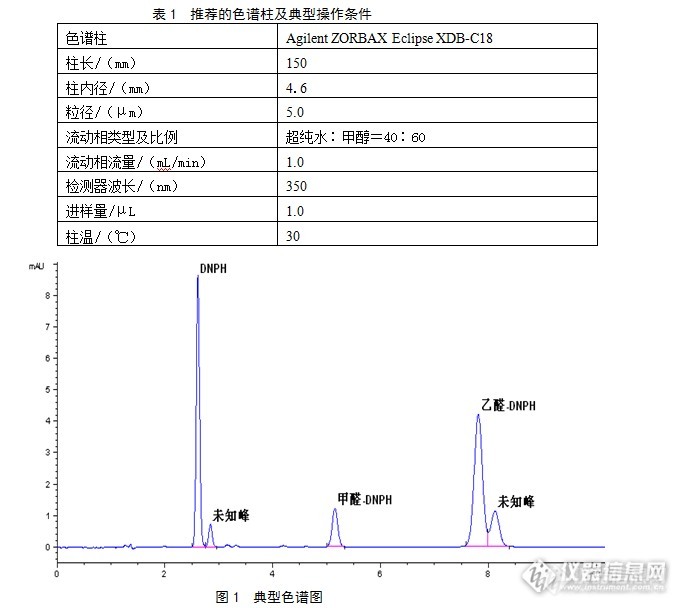
微量甲醛、乙醛液相色谱测定法1 适用范围本标准规定了液相色谱测定水溶液或环氧乙烷样品中的甲醛和乙醛的方法。本标准适用于样品中甲醛、乙醛含量为1~50mg/kg的分析。2 方法概要 液态样品中微量甲醛、乙醛与2、4-二硝基苯肼衍生生成甲醛-2、4-二硝基苯腙(以下称:甲醛-DNPH)和乙醛-2、4-二硝基苯腙(以下称:乙醛-DNPH),在通过固相C18小柱将甲醛、乙醛的衍生物从溶液中萃取出来。随后,使用自动进样器直接将萃取出的甲醛、乙醛衍生物注入[url=https://insevent.instrument.com.cn/t/Mp]气相色谱仪[/url],样品在甲醇和水混合溶液携带下通过毛细管色谱柱分离,使样品里的甲醛衍生物、乙醛衍生物及未反应2、4-二硝基苯肼按照不同顺序先后进入紫外检测器检测。最后,由色谱数据工作站采用外标法得到各物质含量。3 试剂和材料3.1 甲醇:液相色谱(HPLC)纯。3.2超纯水:比电阻率大于18.2ΜΩcm的水。3.32,4-二硝基苯肼(DNPH):分析纯。3.4超声波振荡器。3.5玻璃溶剂过滤器。3.6真空泵。3.7量筒:10mL。3.8量筒:25mL。3.9具塞碘量瓶:250mL。3.10C18固相萃取小柱(SPE)。3.11注射器:5mL。3.12注射器:10mL。3.13烧杯:50mL3.14标准溶液:与分析样品浓度相接近甲醛、乙醛水溶液,自配或外购。4 仪器4.1液相色谱仪:安捷伦科技公司1200型,配有自动进样器、真空脱气、四元梯度泵和紫外检测器,或同类产品。4.2色谱数据工作站:安捷伦科技公司Chemstation工作站,或同类产品。4.3色谱柱:见表1推荐色谱柱,或同类产品。推荐色谱柱典型操作条件见表1,典型色谱图见图1。[img=,681,608]http://ng1.17img.cn/bbsfiles/images/2017/09/201709032112_01_2166779_3.png[/img]5 校正仪器第一次使用或者被改变了某一或某些参数设置(如流量、流动想类型、流动相比例、色谱柱等),应对仪器进行校正。使用已知与样品接近含量的标准甲醛、乙醛溶液与DNPH络合后,在液相色谱仪上分析,测量两物质色谱峰峰面积,计算校正因子k[sub]i[/sub]:k[sub]i[/sub]=标准液中i物质浓度/i物质衍生物峰面积。6 试验步骤6.1用5mL注射器将5mL甲醇注入C18固相萃取小柱,冲洗萃取小柱进行预处理,然后再用5mL超纯水冲洗。6.2分别取10g样品(由于环氧乙烷沸点较低,环氧乙烷产品可直接使用量筒量取11.2mL)、10mL2,4-二硝基苯肼(DNPH)试剂和10mL超纯水转移到碘量瓶内,盖上瓶塞,让混合物反应15分钟。如果观察到沉淀,则应使用超纯水按1:10稀释样品,并按上述描述再次进行操作。6.3使用一个10mL量筒量取10mL的反应混合物,用针筒将其转移到一个6.1中预处理好的固相萃取小柱,注射时应将样品缓慢地推进萃取小柱。然后,使用10mL超纯水重复上述步骤,注意需将所有液体推出萃取小柱。6.4用注射器将8mL甲醇推进萃取小柱,以洗脱富集被固相萃取小柱从样品中萃取出的甲醛-DNPH、乙醛-DNPH及未反应的DNPH溶液。洗脱富集液放置于烧杯中。再向烧杯添加少量甲醇,使溶液的最终重量达到10±0.01g。6.5使用符合4.3中推荐色谱柱或同类色谱柱,采用自动进样器将适量6.4中的洗脱富集样品注入4.1规定液相色谱仪,通过色谱工作站测量各物质出峰面积,利用已测校正因子面积归一定量计算样品中各组成含量。6.6用10mL的去离子水替代样品重复步骤6.2至6.5,测定空白。7 计算7.1溶液甲醛、乙醛含量按下式计算:(外标法)X[sub]i[/sub]=A[sub]i[/sub]×k[sub]i[/sub]式中:X[sub]i[/sub]——被测组分i的含量,mg/kg;A[sub]i[/sub]——被测组分i的衍生物峰面积;k[sub]i[/sub]——被测组分i的绝对校正因子;i——甲醛或乙醛。8 精密度8.1 重复性同一操作者重复测定的两次测定结果的差值应不超过平均值的10%(m/m)。9 结果的表示仪器稳定状态下连续两次测定结果的平均值作为出厂产品的测定结果,生产装置过程控制使用单次测定数据作为结果报告,结果的表示修约至1mg/kg。10注意事项10.1 仪器进样前取样,需摇晃采样容器,以保证分析样品的均匀性。10.2 色谱流动相必须使用液相色谱(HPLC)纯试剂。10.3当分析环氧乙烷或含有环氧乙烷样品时,使用前应事先将试剂、容量瓶和量筒冷却,以防止环氧乙烷受热挥发。10.4 对含有环氧乙烷的样品的任何操作,必须在通风柜中完成,并做好个人防护。
高效液相色谱定量分析1 黄芩苷含量检测(1)色谱条件与系统适应性试验 以十八烷基硅烷键合硅胶为填充剂;流动相:甲醇-水-磷酸(47:53:0.2);检测波长:276nm。柱温:30℃;流速:1mL/min。理论板数按黄芩苷峰计算应不低于4500。(2)测定方法对照品溶液的制备 取黄芩苷对照品适量,精密称定,置棕色量瓶中,加50%甲醇适量,置水浴中振摇使溶解,放置至室温,稀释至刻度,摇匀,制成每1mL含40μg的溶液,即得。 供试品溶液的制备 精密量取样品溶液,置容量瓶中,加水稀释至一定浓度,摇匀,用0.45μm滤头过滤即得。测定法 分别精密吸取对照品溶液10μL与供试品溶液10μL,注入色谱仪,测定,即得。2 高效液相色谱的定量分析药典规定:本品每支10mL含黄芩苷(C21H18O11)计,不少于80mg。本品每1mL含金银花以绿原酸(C16H18O9)计,不得少于0.60mg。(1)黄芩苷高效液相图 见附件1 [img]http://www.instrument.com.cn/bbs/images/affix.gif[/img][url=http://www.instrument.com.cn/bbs/download.asp?ID=101403]附件1 [/url]
求用高速液相色谱法测植物激素含量如题本人想用高效液相色谱法测定花芽中赤霉素的含量想知道实验之前要做怎样的准备工作求助大家谢谢啦
高效液相色谱测定猪组织瘦肉精含量,不知大家用的前处理方法?都来交流下啊







 我要推广仪器
我要推广仪器
 下载APP
下载APP