高分子表征技术专题——基于原子力显微镜的单分子力谱技术在高分子表征中的应用
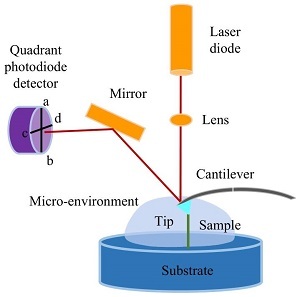
2021年,《高分子学报》邀请了国内擅长各种现代表征方法的一流高分子学者领衔撰写从基本原理出发的高分子现代表征方法综述并上线了虚拟专辑。仪器信息网在获《高分子学报》副主编胡文兵老师授权后,也将上线同名专题并转载专题文章,帮助广大研究生和年轻学者了解、学习并提升高分子表征技术。在此,向胡文兵老师和组织及参与撰写的各位专家学者表示感谢。更多专题内容详见:高分子表征技术专题高分子表征技术专题前言孔子曰:“工欲善其事,必先利其器”。我们要做好高分子的科学研究工作,掌握基本的表征方法必不可少。每一位学者在自己的学术成长历程中,都或多或少地有幸获得过学术界前辈在实验表征方法方面的宝贵指导!随着科学技术的高速发展,传统的高分子实验表征方法及其应用也取得了长足的进步。目前,中国的高分子学术论文数已经位居世界领先地位,但国内关于高分子现代表征方法方面的系统知识介绍较为缺乏。为此,《高分子学报》主编张希教授委托副主编王笃金研究员和胡文兵教授,组织系列从基本原理出发的高分子现代表征方法综述,邀请国内擅长各种现代表征方法的一流高分子学者领衔撰写。每篇综述涵盖基本原理、实验技巧和典型应用三个方面,旨在给广大研究生和年轻学者提供做好高分子表征工作所必须掌握的基础知识训练。我们的邀请获得了本领域专家学者的热情反馈和大力支持,借此机会特表感谢!从2021年第3期开始,以上文章将陆续在《高分子学报》发表,并在网站上发布虚拟专辑,以方便大家浏览阅读.期待这一系列的现代表征方法综述能成为高分子科学知识大厦的奠基石,支撑年轻高分子学者的茁壮成长!也期待未来有更多的学术界同行一起加入到这一工作中来.高分子表征技术的发展推动了我国高分子学科的持续进步,为提升我国高分子研究的国际地位作出了贡献.借此虚拟专辑出版之际,让我们表达对高分子物理和表征学界的老一辈科学家的崇高敬意!基于原子力显微镜的单分子力谱技术在高分子表征中的应用ApplicationofAtomicForceMicroscopy(AFM)-basedSingle-moleculeForceSpectroscopy(SMFS)inPolymerCharacterization作者:张薇,侯矍,李楠,张文科作者机构:吉林大学超分子结构与材料国家重点实验室,长春,130012作者简介:张文科,男,1973年生.分别于1997、2002年在吉林大学化学系(学院)获得学士、博士学位,导师为张希教授;2001~2002年于德国慕尼黑大学(LMU)博士联合培养,导师为HermannE.Gaub教授;2003~2007年于英国诺丁汉大学从事博士后研究.2007年6月至今,吉林大学超分子结构与材料国家重点实验室教授.2011年入选教育部“新世纪优秀人才支持计划”;2015年获得国家杰出青年基金资助.以原子力显微镜及磁镊等技术,从单个分子水平开展超分子作用力及大分子组装结构与组装过程研究,主要研究方向包括:单分子力谱与超分子组装、高分子结晶及力致熔融、核酸-蛋白相互作用、聚合物力化学等.摘要基于原子力显微镜(atomicforcemicroscopy,AFM)的单分子力谱技术以其操作简便、适用面广等优势,成为了单分子领域应用最为广泛的技术之一.本文阐述了该技术的基础原理与实验技巧,包括仪器构造、工作原理、探针与基底的选择、样品固定、实验操作、单分子信号的获得以及数据处理.介绍了基于AFM的单分子力谱技术在合成高分子及生物大分子表征中的典型应用及前沿进展.AFM单分子力谱技术将有助于建立合成高分子的链结构、链组成与单链弹性以及链间相互作用与其宏观力学性能间的关联,帮助理解生物大分子的结构、相互作用与其生物功能之间的联系.AbstractAtomicforcemicroscopy(AFM)-basedsingle-moleculeforcespectroscopy(SMFS)hasbeenusedwidelyintheinvestigationofmolecularforcesbecauseofitsfriendlyuserinterface(e.g.,easytooperateandcanworkinliquid,airandhighvacuumphase)andworldwidecommercialization.ThisreviewisaimedtointroducetheprincipleandprotocolofAFM-basedSMFSincludingthesetup,theworkingprinciple,typicalcurves,thechoiceofAFMtipandsubstrate,immobilizationofsamples,manipulationofthedevice,empiricalcriteriaforsingle-moleculestretchinganddataanalysis.RecentprogressesontheapplicationofAFM-basedSMFSinthecharacterizationofsyntheticpolymersandbiopolymerswerereviewed.Forsyntheticpolymers,theeffectsofprimarychemicalcompositions,sidegroups,tacticityandsolventsonthesinglechainelasticitieswerediscussed.TheapplicationsofAFM-SMFSindisclosingthestructureofunknownmolecule,polymer-interfaceinteractionsandpolymerinteractionsinpolymerassemblies(e.g.,polymersinglecrystal)wereintroduced.Inaddition,thenatureofmechanochemicalreactionsandcharacterizationofsupramolecularpolymerswererealizedviathistechnic.Forbiopolymers,theeffectsofbase-pairnumber,theforce-loadingmode(unzippingorshearing)onthestabilityofshortdouble-strandedDNA(dsDNA)werereviewed.Accordingtothisknowledge,thesingle-moleculecut-and-pastebasedDNAassemblywasthendiscussed.ThetypicalforcefingerprintsoflongdsDNA,proteinsandpolysaccharidesaswellastheforce-fingerprint-basedinvestigationofmolecularinteractionswereillustrated.Finally,theapplicationofAFM-SMFSinrevealingtheintermolecularinteractionsandthemechanismofvirusdisassemblyaswellastheantivirusmechanismoftanninintobaccomosaicviruswerereviewed.Therefore,AFM-basedSMFSisessentialforrevealingtherelationshipbetweentheconformation/compositionofpolymerchainsandmicro/macro-mechanicalpropertiesofpolymermaterialsaswellascorrelatingthemolecularstructure/interactionofbiopolymerswiththeirbiofunctions.关键词AFM单分子力谱 合成高分子 生物大分子KeywordsAtomicforcemicroscopy-basedsingle-moleculeforcespectroscopy Syntheticpolymers Biopolymers 合成高分子材料自诞生以来,迅速地以其优良的物理、化学及力学性能等在军事、航空航天、医疗及其他民用领域得到了广泛应用.其力学性能是最基本、最重要的性质之一,同时受到高分子的单链弹性及链间相互作用的影响[1,2].因此,建立高分子链一级结构、单链弹性及链间相互作用与材料宏观力学性能间的联系,对高分子材料的理性设计至关重要.然而,传统的材料学研究方法,如宏观拉伸实验、X射线晶体衍射、固体核磁及拉曼等技术无论从样品制备到检测均涉及大量分子,体现平均效应,表征宏观力学性能,无法获得单个链或键的性质及行为的相关信息.此外,传统研究方法也无法连续、动态及精确地体现出单个事件的不同步骤(例如高分子在不良溶剂中的塌缩行为),导致很多重要信息无法获取.因此,可在纳米尺度精确操纵与测量的单分子技术,例如基于AFM的单分子力谱,被广泛应用于单个分子的结构、功能及其动态行为的研究中[1~5].利用该技术,人们获得了溶剂、取代基以及立构规整度等因素对高分子单链弹性的影响,验证并改进了一些经典高分子理论模型[1,6~9].该技术还可以研究高分子的构象变化及其在界面的吸附行为,揭示外力诱导下高分子链中化学键类型的变化规律(力化学)[1,10~12].同时,该技术还被用于凝聚态(晶体、层层组装薄膜等)中高分子间相互作用的相关研究[13,14].生物大分子(核酸、蛋白质及多糖等)结构与功能的研究对于认识复杂生命过程的本质,了解疾病的发生发展机制以及开发新型药物与生物医用材料至关重要.因此,AFM单分子力谱技术也被广泛用来研究生物大分子,例如DNA的解链及动态结构变化、蛋白质的折叠与解折叠、生物大分子间的相互作用(病毒的遗传物质与蛋白质外壳的相互作用)等[9,15~20].相关研究深化了人们对这些生物分子所参与的生命过程的认识,并为其功能调控奠定了坚实基础.本文将重点评述AFM单分子力谱技术的基础原理、实验技巧以及该技术在合成高分子及天然高分子领域的典型应用及前沿进展.1单分子力谱的基础原理1.1几种典型的单分子力谱技术迄今为止,诞生了许多单分子操纵技术,例如生物膜力学探测技术、玻璃纤维技术、光学镊子(光镊)、磁性珠技术(磁镊)以及AFM单分子力谱技术[9,21~25].后3种技术的应用较为广泛.光镊利用聚焦激光束产生辐射压力形成的光学陷阱来捕获修饰有样品分子的小球,通过移动激光光束控制小球的移动,实现对样品分子的三维操纵,其时间分辨力能够达到10-4s,被广泛应用于蛋白质折叠及解折叠等研究.但光镊系统构造复杂,对环境要求极高,有效样品捕获率低以及激光束容易对样品造成光和热损伤等不足亟待解决.磁镊技术将样品固定在基底与超顺磁性小球之间,利用外加磁场控制磁球,操纵样品分子,例如旋转等[22].因此,磁镊被广泛用于DNA缠绕及解缠绕等研究中.该技术可以检测低至10-3pN的力值,也被应用于一些极微小力的测量.该技术还能同时对多个磁球进行操纵,实现高通量测试.由于需要通过成像观测磁珠,因而相机的拍摄速度决定了磁镊的时间分辨率,通常在10-2s以上.在众多的单分子力谱技术中,AFM单分子力谱技术的应用最广,理论发展更为成熟[1~5,9,26,27].该技术将样品分子固定在AFM探针与基底之间,通过控制AFM探针的位移来操纵样品分子.该技术具有较高的时间和空间分辨率,较宽的力学测量范围,可以在真空、水相以及有机相等多种环境下工作,因此被广泛地应用于合成与天然高分子等众多体系中的分子内及分子间相互作用的研究.综上所述,光镊及磁镊的力学精度稍高,适用于由弱相互作用及熵弹性所控制的力学性质的研究;AFM单分子力谱更适合较强相互作用或者由焓控制的弹性性质的研究.为了更全面地认识聚合物的结构与力学性质,可以将上述3种单分子力谱技术联合使用.1.2AFM单分子力谱1.2.1仪器构造基于AFM的单分子力谱是AFM的工作模式之一.因此,其基本构造与AFM相同,主要由位置控制系统(压电陶瓷管)、力学传感系统(AFM探针的微悬臂及其顶端针尖)以及光学检测系统(激光二极管、棱镜、反射镜与四象限光电检测器)三部分组成(图1)[9,21,28,29].对压电陶瓷管两端施加电压,可以控制其驱动样品台或AFM探针进行亚纳米精度的位移.z方向的移动用于调整探针与样品间的距离;x,y方向的移动用以调整探针在样品表面的探测位置及范围.光学检测组件中的激光器将激光照射在微悬臂靠近针尖的一端,再反射到四象限光电检测器上.当AFM探针受到样品分子的牵拉发生弯曲时,其反射的激光的位置也会随之变化.据此,可以计算出微悬臂的偏转量,结合微悬臂的弹性系数,可以获得待测样品分子的相关力学信息[3~5].Fig.1TheschematicdiagramofAFM-SMFS.1.2.2工作原理实验前,样品分子的一端通过物理吸附、特异性相互作用或化学偶联等方法被固定在基底.随后,驱动压电陶瓷管使AFM探针逼近待测样品(图2(a)).如果基底对探针没有长程的吸引或排斥作用,微悬臂将处于松弛状态.探针与基底接触后,受力向微悬臂上表面方向弯曲,引起二极管的2个象限间的差分信号(pha-b)的变化(图2(a)与2(b),状态2→3).在此过程中,样品分子会通过化学、物理或特异性作用吸附在探针上,在探针与基底之间形成桥联结构.随后,探针远离基底并恢复松弛状态(图2(a),4),pha-b也恢复初始数值.探针继续远离基底,桥联于探针与基底间的样品分子受到拉伸,导致微悬臂向针尖方向偏转(图2(a),5),引起pha-b的增加(图2(b),5).最后,桥联结构中稳定性最薄弱的部分发生断裂,微悬臂迅速恢复为不受力的松弛状态(图2(a),6),表现为pha-b的突然回落(图2(b),6)[1,9,21,29].每个完整的逼近-回缩过程都会产生pha-b对应压电陶瓷管位移的原始曲线(图2(b))[29].Fig.2(a)SchematicillustrationofthebasicworkingprincipleofAFM-SMFS (b)Originalvolt-piezodisplacementcurves (c)Typicalforce-extensioncurves.Fig.3ElectronmicroscopyimagesofacommercialSi3N4AFMprobe.Fig.4Molecularimmobilizationbasedon(I)physicalabsorption,(Ⅱ)specificbinding,(Ⅲ)gold-thiolchemistry,(Ⅳ)silanizationandenzymaticbiosynthesis.Fig.5Immobilizationofthiol-labeledDNAbasedonsilanizationandbifunctionalPEG.Fig.6Typicalcurvesobtainedinconstantvelocity(a)andforce-clampmode(b),respectively.原始曲线经过校正才能正成为最终的力-拉伸长度曲线(图2(c))[1,2,4,9,21,29].将具有弹性的微悬臂看成弹簧,根据胡克定律F=kcΔx(kc为微悬臂弹性系数,Δx为微悬臂偏转量)可以计算出微悬臂受到的作用力,即样品分子内或分子间的作用力.kc通过对微悬臂在远离基底时热振动所获得的能量谱的积分即可获得;Δx利用图2(b)中斜线部分(状态2→3)的斜率(s),即Δx=s-1pha-b就可以计算出.样品分子的拉伸长度通过从原始数据横坐标记录的压电陶瓷管的位移中扣除Δx获得.至此,pha-b对应压电陶瓷管位移的原始曲线被成功地转化为样品分子的力-拉伸长度曲线.1.2.3力曲线及其含义AFM针尖逼近和远离样品表面的一个循环中可以获得2条力曲线,称为逼近力曲线与回缩力曲线(图2(c))[1,2,4,9,21].逼近力曲线上B区域的形状可以给出样品模量等信息.例如:当AFM探针接触较软的样品时,受到的排斥力随位移缓慢增加;而接触硬度较大的样品时,受到的排斥力快速增加,B区域的力信号与水平基线之间形成近90°的直角.对于回缩力曲线,C-D区域可以给出单分子弹性性质、链结构信息以及分子内、分子间相互作用强度等定量信息.2AFM单分子力谱实验技巧2.1探针与基底的选择AFM探针直接影响力学探测的稳定性、精确度及测量范围[1,2,4,9,21,29].其材质通常是硅或氮化硅,由针尖、微悬臂及承载微悬臂的基片组成(图3).针尖通常是四面体形状,最尖端的曲率半径(tipradius)为几个到几十纳米,高度(tipheight)通长为3~28µm.微悬臂有矩形和三角形2种,长度为7~500µm,厚度为0.5~7µm.其材质及几何尺寸均对共振频率和弹性系数有重要影响,需要根据实验体系来选择探针.对于弱相互作用体系(例如双链DNA的解拉链)[30],应选择相对柔软,即弹性系数小的探针;而强相互作用体系(例如:共价键强度的测量)[31],则需选择相对坚硬,即弹性系数较大的探针.值得注意的是,刚性较大的探针在应力松弛时其内部储存的能量释放速度更快,更适于研究多重键的连续打开与形成的动态过程,例如聚酰胺(PA66)单晶中聚合物链在受力熔融过程中的黏滑运动(stick-slip)[32].此外,一些公司也生产了许多功能化的AFM探针.例如:满足基于巯基-金的化学分子偶联的镀金AFM探针;为了增加激光束在微悬臂上表面的反射率,只在上表面蒸镀金属涂层(铝或金等)的探针等.然而,只存在于微悬臂上表面的镀层,往往导致其上下表面的膨胀系数产生差异,引起热漂移[33].为了减小该热漂移,有些探针只在其微悬臂的尖端进行有限的金属蒸镀(例如MLCT-BioDC型号探针).如需增加时间分辨率,可以选用超短探针[34].但超短探针的弹性系数通常较大.科研人员曾利用离子束刻蚀的方法将微悬臂做成镂空结构,同时保证了时间分辨率和弹性系数[35].然而,使用较小尺寸微悬臂时,激光容易“漏射”到样品表面,发生反射,与微悬臂表面的反射光产生干涉,导致力曲线出现大幅度波动.为了减少这种干涉效应,通常可以采取以下几种策略:(1)减小汇聚到微悬臂表面的激光光点的大小,从而减小漏光;(2)选用横向尺寸较大的微悬臂,增大反射面积;(3)选择透明基片(例如玻璃片)固定样品,降低基片的反射率;(4)适当增加样品平面相对于微悬臂平面的角度,降低反射光的相干性.AFM探针需要被牢固地固定在夹具上,以减少系统漂移.为了提高微悬臂检测的灵敏度,将激光光斑尽可能地照射在微悬臂的最前端.仪器调试完毕,让整个系统平衡10~30min,使微悬臂上下表面材质差异所引起的界面张力达到平衡,减小系统漂移.如在同一个样品上进行力谱探测的时间较长,且实验前期及后期微悬臂反射到光电检测器中的激光强度(sum值)有较大变化,可以在实验开始及结束时分别校正微悬臂的光学灵敏度及弹性系数.如数值差异较大,则实验前期与后期所得数据分别用初期及实验结束时的校正参数进行转换分析,防止由悬臂弯曲等因素导致激光点位置偏移,进而影响测试结果.AFM单分子力谱实验中常用基底(样品固定用)主要有石英、玻璃、硅片、云母及镀金片等[1,4,9,21,29].硅片较为平整,容易通过物理或化学修饰固定样品,最为常用.玻璃和石英片的平整度不如硅片,但其较好的透光性可以同时进行光学成像和单分子力谱实验.同时,透明基片有助于减小由较大的激光光斑或较小的微悬臂所产生的干涉现象.对于细胞类样品,也可直接在接种了细胞的聚苯乙烯培养皿上进行单分子力谱实验,以弥补玻璃基底细胞黏附率低的不足.含有巯基的样品分子可以选择表面有金镀层的基底.但金镀层反射率较强,当微悬臂较小时,漏到金片表面的激光较多,容易产生干涉现象.除了前述4种消除干涉的策略,还可以通过差减拉伸力曲线与松弛力曲线的方法来扣除干涉条纹[36].总之,要根据体系选择合适的基底.2.2样品固定与偶联成功进行单分子力谱实验的关键环节是样品分子在AFM针尖与基底之间的有效偶联,形成桥联结构,即样品分子的固定.根据相互作用本质的不同,样品固定方式可以分为物理吸附,特异性相互作用以及化学偶联(图4)[1,20].物理吸附主要利用样品分子与基底或针尖间的物理相互作用,例如范德华力、氢键、静电相互作用以及疏水作用力等,吸附样品分子(图4,Ⅰ)[9,21,26,27].通常将待测样品配成稀溶液,滴加到固体基片表面,吸附一定时间后,润洗掉游离及吸附不牢固的分子即可.为了增加单分子探测的几率并减小多分子信号的干扰,可以降低样品溶液浓度、减少吸附时间,以降低样品分子的吸附密度[2,28].对于吸附能力强且分子在溶液中易发生聚集的样品体系,需要配制极稀溶液来实现单分子拉伸探测[41].基于物理吸附的样品,制备虽然操作简便、快速,但由于探针在目标分子上的接触位置无法控制,导致拉伸长度及断裂力值随机分布.其次,物理吸附的作用力通常较弱且不可控,不适用于强作用力体系的研究.但在有些样品体系中,例如:拉伸多糖分子时,物理吸附也可以达到较高的断裂力(如1nN左右)[1,37~41].该现象主要源于样品分子在基底及AFM探针表面的多点物理吸附[1,37,41].此外,当探针以较大的下压力(例如2nN)接触样品分子时,在力诱导下样品分子有可能与AFM探针发生力化学反应,形成共价键,从而增大断裂力[1,37,42].特异性相互作用也经常用于固定样品分子(图4,Ⅱ).该方法专一性强、结合强度确定.其中生物素-链霉亲和素(biotin-streptavidin)系统(亲和常数高达1015mol/L)是最常用的分子对[9,21,26,27,43].此外,谷胱甘肽-谷胱甘肽S转移酶[44]以及镍离子(Ni2+)-氮川三乙酸(NTA)-组氨酸标签(His-tag)[45]的配位作用等也可以用于目标分子的固定.特异性分子对可以通过简单的化学反应被修饰在样品分子的末端或AFM探针、基底的表面,也可以通过基因工程方法被构建在蛋白质类样品的末端.上述分子对的相互作用强度通常在100pN左右,断裂力值高于该数值的体系不能采用特异性相互作用固定样品分子.样品分子还可以通过化学修饰直接固定在AFM探针或基底上(图4,Ⅲ与Ⅳ).化学修饰通过形成共价键固定样品分子.其修饰过程比物理吸附复杂,但形成的共价键较为牢固,断裂力可达纳牛顿(nN)级别,适用的力值测量范围较大.基于金-硫(Au-S)相互作用的化学偶联是常用的化学修饰方法之一(图4,Ⅲ)[21,26,27].该体系需要向待测样品中引入巯基.蛋白质类样品可以通过基因工程方法引入侧链含有巯基的半胱氨酸;核酸或高分子样品,可以通过化学方法在样品合成时直接引入巯基.随后,选用镀金的探针或基底,即可通过Au-S相互作用固定样品.研究表明金表面的氧化还原状态、溶剂的种类(水或乙醇)、反应环境的pH值以及反应时间等均影响目标分子在金表面偶联的稳定性[46].硅烷化是另一种常用的化学偶联方法(图4,Ⅳ)[9,21,26,27].该方法首先利用食人鱼(Piranha)洗液(V(98%H2SO4)∶V(30%H2O2)=7∶3)或者紫外臭氧等方法处理AFM探针或基底,使其表面羟基化.随后利用末端修饰有活性基团(例如氨基、羧基、巯基、环氧基等)的硅烷偶联剂与上述羟基反应形成硅氧键.最后,使探针或基底引入的活性基团与样品分子中相应的官能团反应形成共价键,从而固定样品分子.含有3个可水解基团的硅烷化试剂(例如三甲氧基或三乙氧基硅烷)与羟基反应效率较高.但在少量水存在时,该试剂可以发生分子间缩合,生成低聚硅氧烷,不仅对力谱实验产生干扰,还会增加样品分子的修饰密度,产生多分子信号.为了避免上述现象,可以选用只含有一个可水解基团的硅烷化试剂[46].末端含有氨基的硅烷化试剂在单分子力谱实验中的应用较为广泛.该氨基可以与含有琥珀酰亚胺羟基酯(NHS)的双功能偶联剂进一步反应固定目标分子(图5)[47],也可以在EDC(DCC)/NHS的催化下与羧基反应形成酰胺键来实现偶联.近年来,研究人员将硅烷化反应与基于酶催化反应的化学偶联结合,固定蛋白质类样品(图4,Ⅳ)[48].该方法利用氨基硅烷化试剂修饰探针或基底,再利用氨基巯基交联剂引入末端含有巯基的桥联蛋白(例如半胱氨酸).桥联蛋白的另一端含有甘氨酸-亮氨酸(GL),利用可快速催化GL与天冬酰胺-甘氨酸-亮氨酸(NGL)间连接反应的OaAEP1酶,就可将末端含有NGL的蛋白样品固定在基底或探针上.半胱氨酸、GL以及NGL均可以通过基因工程方法引入桥联蛋白及目的蛋白末端.通过共价键固定的样品体系,可以将双官能团偶联剂与惰性(单官能团)分子偶联剂(例如另一个末端是甲基的硅烷化试剂或者硫醇分子)共同修饰到基片表面(图5)[1,49].惰性偶联剂不含与样品分子反应的活性基团,无法连接样品分子,因此可以稀释样品分子,增加单分子信号[3].研究相互作用力的体系,例如受体与配体相互作用,柔性的PEG连接分子通常被引入样品分子与探针或基底之间(图4,Ⅱ)[9,21,27].连接分子主要有以下几种作用:(1)减小样品分子在探针或基底的非特异性吸附;(2)使样品分子构象自由、取向灵活,在相互作用的过程中结合得更加充分;(3)将断裂信号平移到更长的断裂长度,减小非特异性相互作用对目标力信号的干扰.图5给出了基于硅烷偶联剂和带有双官能团PEG的单股DNA共价固定方法.样品分子与AFM针尖的偶联方式决定了实验过程中一些实验参数的设置.依靠物理吸附的体系,探针接触样品表面时需要施加较大的下压力(例如1nN)和较长的停留时间,以增大样品分子在针尖表面吸附的概率及稳定性.利用特异性相互作用或化学反应的偶联,探针接触基底时则需采用尽可能小的下压力和较短的停留时间,避免破坏针尖表面修饰的有限数量的功能基团.此外,对于化学修饰的AFM探针,其表面的功能基团可能随拉伸次数的增加而变化,影响实验效率,因此需要适时更换探针.此外,如果力谱实验中多分子信号频繁出现,可以采取如下策略补救:(1)降低探针作用在样品上的下压力及停留时间,减小样品分子与探针间的作用力;(2)探针远离基底时,首先使探针在z方向小范围移动,使较短的分子率先被拉伸,随后逐渐增加探针的移动范围,使较短的分子先从探针上脱落,在探针与基底间保留单个、较长的分子;(3)采用“飞鱼”模式来获取单分子信号[1],即控制探针在基底上方一定距离内程序性地靠近-远离基底,而不直接接触,当探针接触到一端固定在基底而另一端自由运动的样品分子并成功偶联后,再对该分子进行拉伸.2.3恒速与恒力模式恒速模式是AFM单分子力谱常用的测量模式(图6(a))[9,21,26,27,29].该模式下,压电陶瓷管控制AFM探针以恒定的速率对样品分子进行拉伸操纵,可以直接探测分子内或分子间作用力的强度、分子链长度等信息.获得的典型力学信号通常有单峰、锯齿峰或平台等.如果样品分子与探针及固体基片间的桥联结构足够稳定,还可以对同一个样品分子进行往复拉伸-松弛操纵,获得分子内或分子间的相互作用以及相应结构的动态破坏与形成过程[50].对于非平衡体系,还可以获得不同拉伸速率下,体系内力的强度随力加载速率变化的相关信息,获得体系解离速率常数及能量转换路径的信息[51].在恒速模式的基础上还发展了ForceMapping方法,即在样品表面选定范围内的不同位点进行恒速拉伸[9,21,27].该方法可以探测样品表面不同区域的黏附力情况,也可以研究细胞膜表面蛋白质的分布情况等.AFM单分子力谱另一种常用模式是恒力模式,也称力钳(force-clamp)模式(图6(b))[52~54].该模式通过力反馈系统精确控制压电陶瓷管的位移,使施加在样品分子上的作用力保持恒定,记录分子链长度随时间的变化过程,被广泛应用于蛋白质解折叠等领域[34].以多聚串联蛋白质解折叠实验为例[34],先将蛋白质快速拉伸到接近其解折叠力的力值附近,保持外力恒定,一段等待时间后,一个折叠结构在外力作用下打开,释放出一定的长度.此时,探针施加在蛋白质上的力快速下降,仪器反馈系统快速响应,压电陶瓷管再次拉伸蛋白分子到设定力值,维持恒力模式.该过程表现在长度-时间曲线上是长度的突然增加,代表蛋白解折叠所释放的长度.拉伸长度在下一个解折叠事件前保持恒定.随后,其余折叠结构依次打开,最终得到台阶状的长度-时间曲线(图6(b)).从上述曲线中可以获得解折叠路径(包括中间态)及折叠结构寿命的信息(相邻台阶出现的时间间隔).恒力模式还可以研究蛋白质解折叠的逆过程,即折叠过程.将施加在解折叠蛋白分子上的外力突然减小到较低的力值(如十几个皮牛)并保持恒定,就可以原位跟踪蛋白质的动态折叠过程.值得指出的是,为了准确跟踪检测蛋白解折叠等动态结构变化信息,真实描述相关过程,仪器的力学反馈系统需要足够快.2.4单分子信号的指认AFM单分子力谱实验的核心目标是获得单个分子的拉伸信号.因此,指认单分子信号、规避或消除体系中的多分子信号至关重要.单分子信号的指认是一个有挑战性的任务,人们总结了一些半经验性的标准用于单分子信号的指认[1].首先,可以统计体系的信号率,如果信号率较高(10%),则预示体系内可能存在着一定比例的多分子拉伸[1].随后,可以观察单条力曲线上力信号的个数、形状及力值.通常,单分子拉伸只对应力-拉伸长度曲线上一个单独的峰.如果力曲线上出现多个连续、大小不一的峰或平台,则可能对应着多分子拉伸[49].其次,可以对数据归一化处理[1].同一实验体系中,相同结构与性质的分子受到的拉伸力与被拉伸的长度成正比.据此可以对不同拉伸长度的曲线进行归一化处理(拉伸长度除以完全伸展长度).如果归一化曲线能完好的重合,则证明该批数据均来自单个分子的拉伸操纵.此外,往复拉伸实验中,如果分子链内部不存在高级结构及强相互作用,即使经过多次的拉伸、松弛操纵,单分子的拉伸及松弛力曲线依然可以重合.除了上述方式,还可以利用一些模型拟合力曲线,比较拟合参数(如库恩长度,相关长度或者链段弹性系数等)来甄别单分子数据[1].如果拟合参数相差较大,则证明存在多分子拉伸信号.常用的模型主要有蠕虫链模型(WLC)与自由连接链模型(FJC)[1,2].WLC模型主要用于描述刚性及半刚性高分子.FJC模型中高分子弹性的变化主要来自熵的贡献,主要用于柔性高分子以及刚性高分子低力值区域的单分子力谱数据的拟合.例如:研究人员将相互作用的MUC1多肽与scFv抗体通过PEG连接分子分别修饰在探针与基底上,进行单分子操纵.利用FJC模型拟合数据后,可以准确地区分出1~6个分子对被拉伸时分别对应的力曲线[3,55].此外,已知聚合物的力学指纹谱也可以被用来指认单分子拉伸信号[46].PEG、羧甲基化淀粉以及多聚蛋白质的力学指纹谱是被经常采用的单分子拉伸指示剂.为此,可以将待测分子与已知指纹图谱的分子进行串联(图7)[49].需要注意的是待测体系的力学稳定性要大于内标分子产生力学指纹谱所需的力值.Fig.7Basicstrategytoisolate/identifysinglechain/moleculepairstretching.2.5力谱数据的分析处理单分子力谱数据可以给出的信息包括长度及力值的定量信息.为了更精确地描述这些定量信息,通常需要对大量力学信号进行统计分析[1].常用的统计方法是将所得数据以柱状图形式呈现,进行高斯拟合,得出最可几值.此外,还可以利用自由连接链模型及蠕虫链模型对数据拟合,获得库恩长度、相关长度或者链段弹性系数等信息[1].近年来,这些经典模型不断被修正,应用范围逐渐被拓展[56].例如:FJC模型中了增加参数Ksegment,表征高分子链中每一个链段的弹性,被修正为可伸长的FJC模型(eFJC).该模型中,每一个链段类似弹簧,受力过程中伸长,可以更加精确地描述高分子受力时的弹性行为.为了更好地描述高分子主链的固有弹性,即本征弹性,由量子力学(QM)计算得到的非线性单链焓弹性模量被整合到WLC、FJC及FRC模型中,得到了QM-WLC、QM-FJC与QM-FRC模型[57].在特定情况下,如水环境或真空条件,侧基和环境的非共价相互作用会对高分子链弹性产生影响.为了得到上述情况下高分子主链的弹性,基于两态(two-states)系统的非共价作用动力学被引入,创建了TSQM-WLC、TSQM-FJC及TSQM-FRC模型.上述修正模型能够更加精确地定量高分子链的结构及性质[57].一些非平衡态体系,例如受体配体的解离、力诱导下的转变等,力加载速率会影响力-拉伸长度曲线的形状.因此,可以在较大力加载速率范围内,观察上述非平衡体系的力强度变化,获得动态力学谱[1].之后采用诸如Bell-Evans模型等对其进行拟合,分别可以得到沿着受力方向的结合状态与过渡状态间的距离(xβ)以及体系没有外力作用下的解离常数(Koff).3AFM单分子力谱的典型应用3.1合成高分子体系3.1.1聚合物单链弹性聚合物主链结构对其单分子弹性性质具有重要影响.研究人员利用AFM单分子力谱详细地研究了导电高分子聚苯胺(PANI)在氧化、还原及掺杂状态下的单链弹性性质(图8)[58].实验结果显示这些聚苯胺的力曲线呈现出类似变化规律:力值随着拉伸长度的增加呈单调上升.利用FJC模型拟合上述力曲线,结果表明聚苯胺分子氧化态时刚性最大,掺杂状态时最为柔软.这是不同状态的聚苯胺分子中芳香胺与醌二亚胺的含量不同所导致的.Fig.8Normalizedforce-extensioncurvesofPANIindoped(red),oxidized(blue),andreduced(black)states,respectively(ReprintedwithpermissionfromRef.[58] Copyright(2009)AmericanChemicalSociety).除了主链的氧化还原态外,聚合物主链的立构规整性对其弹性也有重要影响(图9)[1,9].在甲苯溶液中拉伸反式-及顺式-聚异戊二烯时,前者在低力值区体现出较大刚性,而后者则表现出明显的熵弹性.FJC模型的拟合也验证了顺式聚异戊二烯较高的熵弹性.这是由于反式聚异戊二烯以伸展的构型存在,而顺式聚异戊二烯则是扭曲构型.相同外力作用下,顺式聚异戊二烯构象数变化较大,熵弹性较大.该研究表明即使反式聚异戊二烯处于溶液中这种高弹态,其熵弹性仍然不如顺式聚异戊二烯强.Fig.9Normalizedforce-extensioncurvesofcis-andtrans-poly(isoprene)intoluene(ReprintedwithpermissionfromRef.[1] Copyright(2003)ElsevierLtd.).AFM单分子力谱还被用于探究高分子侧链结构对其弹性行为的影响[1,2,9,59].聚丙烯酰胺(PAAM)与聚N-异丙基丙烯酰胺(PNIPAM)主链结构一样,前者重复单元侧链只含有2个氢原子,而后者则含有异丙基与氢原子.这种分子结构的差异导致2种高分子的水合能力不同.水溶液中,PAAM在低力值区域表现出明显的熵弹性.PNIPAM由于侧链异丙基体积较大,更加伸展,熵弹性没有PAAM显著,但在高力值区域表现出明显的焓弹性,如图10所示[1,59].Fig.10Normalizedforce-extensioncurvesofPNIPAMandPAAMinwater(ReprintedwithpermissionfromRef.[1] Copyright(2003)ElsevierLtd.).人们又在单分子水平研究了侧链树枝代数对主链重复单元为苯乙烯与马来酸酐的聚合物GnMA-g-BA(n=1,2,3)弹性的影响[60].在四氢呋喃中拉伸时,G2MA-g-BA分子的刚性大于G1MA-g-BA,表明侧链树枝代数的增加使高分子体积变大,导致其刚性增强.然而,侧链树枝代数增加的同时也导致主链周围空间效应的增大,从而减小酰胺侧基与溶剂间的氢键相互作用.因此,在三氯甲烷中实验时,G2MA-g-BA的刚性反而弱于G1MA-g-BA.故考察聚合物侧基对单链弹性的影响时需要注意考虑溶剂化效应.除了聚合物链本身的组成结构会影响其弹性性质外,聚合物所处的环境(如溶剂)也会影响高分子弹性.例如:高分子在良溶剂中通常以无规线团形式存在,而在不良溶剂中则塌缩成小球[6,61,62].研究人员利用AFM单分子力谱研究了聚苯乙烯(PS)链段在不同溶剂中的弹性行为.在环戊烷中拉伸PS分子时,由于环戊烷破坏了间隔苯环之间的π-π堆积,PS本征弹性能够得以体现[63].然而,在不良溶剂中拉伸形成塌缩小球的PS时,则得到了带有平台与单峰的力曲线(图11(a)与11(b)).平台对应着塌缩结构在外力诱导下逐渐被打开的过程,单峰则对应着伸展后的PS分子在外力拉伸下的弹性行为.利用WLC模型模拟上述单峰信号.假设塌缩小球内部的高分子链段是松弛的,则拟合曲线的上部面积代表从塌缩结构变成伸展状态的过程中高分子与溶剂的界面能的增加,即PS的疏水塌缩过程由界面能支配(图11(c))[61].随后,PS纳米粒子的疏水水合自由能与自身尺度间的关联也被深入探究[64].当PS纳米粒子的尺寸1nm时,水分子在其表面重排,体系发生焓变,水合自由能则与其表面积呈线性关系.Fig.11(a)IllustrationofAFM-SMFSstudyoftheglobule-to-coiltransitionofPSinwater (b)Theforce-extensioncurve(red)ofPSwithentropicelasticstretchingportionfitbytheWLCmodel(blue) (c)Thermodynamicrelationshipsbetweenpolymersandfreemonomersinvariousconformationalstates(ReprintedwithpermissionfromRef.[61] Copyright(2012)AmericanChemicalSociety).通过AFM单分子力谱获得的样品弹性性质还可以用于推测其结构,因此研究人员尝试利用该技术探索未知分子.例如:无定形红磷的分子结构无法通过传统的研究方法获得,其结构一直未知.因此,研究人员将红磷吸附在玻璃基底上,利用AFM探针在乙醇中拉伸红磷分子[65].如图12所示,实验获得的力曲线与拉伸链状高分子类似,表明红磷为线性高分子,并且其平均表观链长约为106nm.利用QM-WLC模型进行理论计算后发现,实验获得的红磷拉伸的力曲线与一种“之字形”梯状的拟合曲线高度重合.因此,无定形红磷可能为“之字形”梯状的线形高分子.Fig.12Theforce-extensioncurveofa-redP(solidblackcurve)andtheQM-WLCfittingcurves(dottedlines)ofitspossiblestructures(inset),respectively(ReproducedwithpermissionfromRef.[65] Copyright(2019)ofWiley-VCHVerlagGmbH&Co.KGaA,Weinheim).3.1.2高分子-界面相互作用高分子在界面的吸附形态对于很多领域(例如界面黏附调控以及层层组装膜构筑等)非常重要,但通常难于表征.AFM单分子力谱被成功用于研究高分子在界面处的吸附形态及解吸附力[1,9].研究表明,高分子在界面处的吸附形态影响着力曲线的形态(图13(a)).例如:通过共价键被修饰在硅基底上的聚二甲基硅氧烷(PDMS)分子,解吸附时产生了带有单峰的力曲线.这表明该分子是以大的“环状”结构(bigloop)或“尾部”结构(tailstructure)吸附在基底.聚四乙烯基吡啶(PVP)在甲醇溶液中通过氢键吸附在氨基修饰的基底上,解吸附时得到了带有锯齿形力信号的力曲线.这意味着PVP分子是以不规则的小的“环状”结构(loopstructure)吸附在基底上,吸附位点较多,力曲线上每一个锯齿峰均对应着环状结构依次从基底上解吸附的过程.然而,当聚乙烯甲酰胺(PVAm)分子通过静电作用力吸附在基底,其解吸附的过程则获得了带有长平台的力曲线.这表明PVAm是以“平躺”形态(train-likestructure)吸附在基底.上述结果表明AFM单分子力谱是研究高分子界面吸附的有效方法.Fig.13(a)Force-extensioncurvepatterns(toprow)andthecorrespondingadsorptionconformations(bottomrow)(ReproducedfromRef.[1]withpermissionofElsevierLtd.) (b)SchematicdrawingofthebreakageofAu-AubondsmeasuredbyAFM-basedSMFS (c)Thestrengthofthiol-goldcontactsonoxidizedandreducedAusurface(ReprintedwithpermissionfromRef.[46] Copyright(2014)NaturePublishingGroup).AFM单分子力谱还被用于研究末端含有巯基的聚合物与金表面的相互作用(图13(b))[46].首先,巯基通过PEG连接分子被固定在AFM探针上,然后接近金基底.Au-S键形成后,再控制探针远离基底.在还原金表面实验获得的断裂力的最可几值约为0.6nN.为了验证该力值是来源于Au-S键的断裂还是Au原子被从基底拉出所导致的Au-Au键断裂,研究人员将上述实验中得到的修饰有金原子的AFM探针继续在修饰有巯基的基底上进行实验,结果得到了类似的断裂力值.这证明,施加外力时,Au-S结合位点处的Au-Au键发生断裂,Au原子被拉出.结合XPS与拉曼光谱技术,人们发现巯基与金相互作用过程中首先形成配位键,而后转变为共价键.同时,氧化金表面及较长的反应时间能增强巯基与金形成的结合位点的力学稳定性(图13(c))[46].为了研究金与其他硫族元素相互作用的规律,研究人员分别合成了含有不同硫族元素的嵌段聚合物(PEG-PUX-PEG(X=S,Se,Te)),研究了该聚合物链在金表面的吸附形态及强度[10].结果显示该种嵌段聚合物以规律的小的“环状”结构(loopstructure)吸附在基底上,且结合位点较多,因此实验得到带有规则锯齿形力信号的力曲线.4nm的锯齿间距与聚合物链中相邻硫族元素间的链段长度相一致.对断裂力值的分析表明Au-Te键的强度最强,Au-Se键次之,Au-S键最弱[10].这证明相对原子质量大的硫族元素与金形成的键强度较大.此外,人们还利用AFM单分子力谱研究了聚多巴胺与氧化钛基底的相互作用[66],以及高分子在固液界面的摩擦行为的本质[67].3.1.3高分子组装体中分子间相互作用半结晶性高分子广泛存在,其在高分子材料中占据非常大的比例.高分子结晶相的结构对材料的力学及光电性质等诸多性质具有重要影响.从单分子水平研究高分子链结构对结晶过程、晶体结构以及材料力学性质等的影响规律,对于进一步揭示结晶机制、建立链结构与材料性能之间的联系、设计发展高性能高分子材料具有重要意义.然而相关研究充满挑战,通过将原子力显微镜(AFM)成像与单分子力谱技术有机结合,并辅以合适的样品制备及偶联方法,研究人员成功地将单条高分子链从聚氧乙烯(PEO)晶体中提拉出来,定量测量了晶体中高分子链间作用力的大小(图14(a)与14(b))[13].Fig.14(a)SchematicillustrationofAFM-basedSMFSstudyonaPEOsinglecrystal (b)TypicalforcecurvesrecordedintheAFM-basedSMFSexperimentonagold-nanoparticle-labeledPEOsinglecrystal(ReprintedwithpermissionfromRef.[13] Copyright(2011)AmericanChemicalSociety) (c)SchematicillustrationofthehydrogenbondchangeinPA66singlecrystalwhenslippingonerepeatunitalongthepullingdirection (d)TheforcecurvesofPA66obtainedduringtheunfoldingofafoldedstructure(ReprintedwithpermissionfromRef.[32] Copyright(2018)AmericanChemicalSociety).在此基础上,研究人员进一步克服样品制备等难题,将其发展成一种普适性的研究方法,成功地将该方法应用到聚乙烯(PE)、尼龙66(PA66)、尼龙6(PA6)、聚乳酸(PLLA)和聚己内酯(PCL)等众多高分子晶体体系当中,结合聚合物单分子力谱和受控分子动力学模拟,系统考察了晶体中聚合物链构象(螺旋、平面锯齿和非平面锯齿)链组成及外界环境等对纳米力学性质的影响规律:(1)发现晶体中高分子链运动模式的纳米力学特征,螺旋链在受力拉伸时采取螺旋运动模式,力值变化较平稳、波动较小,而锯齿链会发生黏滑运动(stick-slip)导致力值出现锯齿状波动[68,69];同时首次通过单分子实验观测到PA66及PA6晶体中高分子链在受力形变过程中多重氢键的动态断裂与重新形成的粘滑运动过程[32](图14(c)与14(d));发现高分子链在晶体中滑动过程中进行高速旋转直至氢键重新形成;结果还显示,stick-slip运动既受主链结构及链折叠模式的影响,也受力加载装置的弹性系数、无定形区域的链长度等因素的影响;例如:弹性系数较大的探针在松弛过程中储能释放速度较快,链上的残余应力较小,因此在链段滑移后有利于氢键再次形成,表现为力曲线上锯齿峰的数量较多(图14(c)).基于上述具有指纹特征的力谱,研究人员成功地发展了高分子晶体中链折叠模式的定量化研究方法,该方法无需对样品进行标记,具有很好的普适性,研究结果显示溶液相制备的高分子晶体中近邻规整折叠模式所占比例很高(≥91%),表明分子内相互作用对结晶过程起到了关键作用[70].除了晶体,层层组装膜中聚电解质分子间的相互作用及其结合/解离的动力学性质也受到了关注.为此,聚阴离子电解质形成的薄膜被固定在基底表面,聚阳离子电解质PM2VP249被修饰在AFM探针上,进行单分子力谱实验(图15)[14].结果显示,电荷相反的电解质有2种结合模式:一种是在膜的表面发生表面沉积(图15,I),另一种是进入到膜的内部进行内部包埋(图15,Ⅱ).内部包埋模式受到盐离子浓度的影响较大.Fig.15Schematicdescriptionofthestrengthmeasurementofionicbondsandthetypicalforcecurvesrecordedduringthedisruptionofazipperofionicbonds(I:oneatatime Ⅱ:simultaneousdisruptionofcooperativecomplexeswithmultiplebonds)(ReprintedwithpermissionfromRef.[14] Copyright(2012)AmericanChemicalSociety).3.1.4力诱导化学反应近年来,力响应功能基团(亦即力色团),即受到外界刺激时能够发生特定键断裂的功能基团,受到人们的关注[71~78].这些力色团可以赋予材料更高的韧性、自修复性等,并可用于材料体系内部应力损伤的探测.定量研究高分子中的化学键在外力作用下的断裂及转变,对于揭示力化学反应的本质、开发新型力色团继而实现高分子材料的增强增韧以及内部应力大小的可视化呈现具有重要意义[11,31].人们利用AFM单分子力谱揭示了一系列含有力响应基团的高分子在外力诱导下的化学反应的本质与特点[72,79~92].例如带有多个重复四元环功能基团的肉桂酸类高分子被共价桥联在AFM探针与基底之间进行拉伸操纵(图16)[11],获得了带有锯齿峰平台的力曲线.每个小锯齿的峰间距约为2.4nm,与打开一个和四元环相连的冠醚分子所增加的2.46nm的长度极其相似(图16).因此,力曲线上每一个小锯齿峰都代表着一个四元环开环以及与之相连的冠醚分子链的应力释放过程.锯齿峰的高度则对应着四元环开环反应所需要的力值.实验中获得了力值约为1nN(粉色区域)与2nN(蓝色区域)的2种锯齿峰,分别对应顺式及反式四元环开环所需外力的大小(图16).该结果表明,即使是同样的力色团,其在主链上的不同连接方式也可以导致其力学性质的不同,为材料力学性质的调控提供了新思路[11].Fig.16Arepresentativeforce-extensioncurveofcopolymerP2(ReprintedwithpermissionfromRef.[11] Copyright(2017)NaturePublishingGroup).马来酰亚胺-巯基的化学反应常常被用于蛋白质的偶联或修饰中.然而,由于反米歇尔加成反应及巯基交换的原因,马来酰亚胺-巯基交联产物的稳定性很差.但如果上述交联产物能够水解开环,其稳定性将被大幅度提高.因此人们在单分子水平探索了机械力对马来酰亚胺-巯基键稳定性的影响[31].结果表明,在水溶液中断裂马来酰亚胺巯基键(S―C键)需要大约1nN的作用力,而在乙腈中只需要0.4nN.这是由于外力作用下,马来酸硫醚在水溶液中发生水解开环与反米歇尔加成反应,而在乙腈中只发生后一种反应.不受力的情况下,马来酸硫醚的水解开环反应速率很慢,半衰期约为100h.而在外力诱导下,其水溶液中的开环反应速率被提高至1s左右.因此,通过外力诱导引起开环反应而提高了马来酰亚胺-巯基键的稳定性.该项研究提供了一种通过施加外力来稳定马来酰亚胺-巯基键的方法.3.2超分子聚合物3.2.1超分子聚合物的表征超分子聚合物的非共价连接和动态性在赋予其独特性质的同时,也使其分子量等表征变得十分困难.AFM单分子力谱被成功用于具有一定稳定性的长链超分子聚合物的表征[93,94].例如:研究人员曾利用2-脲基-4-嘧啶酮基团(UPy)修饰AFM探针与基底,然后向体系中加入UPy-UPy溶液,依靠超分子组装在探针与基底之间形成超分子聚合物的桥联结构.根据拉伸曲线上的断裂长度以及单个Upy的长度,可以计算出超分子聚合物的聚合度信息(断裂长度/Upy单元长度)(图17(a))[95].除了用于这种基于多重氢键的超分子聚合物体系外,AFM单分子力谱技术还被成功用于客体增强的超分子聚合物体系的研究.该实验中科研人员利用溴化吡啶衍生物DADV分子间的电荷转移相互作用,再结合葫芦脲CB对该复合物的主体增强相互作用,使CB加载DADA,形成超分子聚合物(图17(b))[96].在对该超分子聚合物的拉伸操纵过程中得到了带有一个单峰或者平台状力信号的力曲线(图17(c)).平台力信号对应着超分子聚合物从基片表面的解吸附过程.统计结果显示,该超分子聚合物的最可几分子长度为60nm.此外,人们还合成了首尾均含有蒽的单体,利用π-π相互作用形成超分子聚合物.单分子操纵实验显示,链段长度为23.9nm.以上结果表明AFM单分子力谱技术是表征超分子聚合物的有效手段.当然,如果超分子聚合物的力学稳定性过低,将导致该聚合物链被完全拉伸之前就已经发生断裂,则无法给出聚合物的信息.Fig.17(a)Arepresentativeforce-extensioncurveofUPybasedsupramolecularpolymers(ReprintedwithpermissionfromRef.[95] Copyright(2005)Wiley-VCHVerlagGmbH&Co.KGaA,Weinheim) (b)Illustrationofthesupramolecularpolymerformationbasedonmultiplehost-stabilizedcharge-transferinteractions (c)Force-extensioncurvescorrespondingtothedesorptionofsupramolecularpolymerchainsfromthesubstrateinthesolutionofDADV-2CB[8]andthehistogramofthelengthofplateausontheforcecurves(ReprintedwithpermissionfromRef.[96] Copyright(2010)Wiley-VCHVerlagGmbH&Co.KGaA,Weinheim).3.2.2动态力学性质研究近年来,超分子聚合物的动态力学性质成为了研究的热点,因为该性质与材料的刺激响应性以及自修复等性质直接相关.AFM单分子力谱已被成功用于研究聚[2]索烃中索烃大环的受力运动规律(图18(a))[97].实验获得了带有锯齿峰的力曲线,对应着氢键锁定基元的连续打开过程.对同一条聚合物链进行往复拉伸过程中,拉伸与松弛曲线上都可以获得锯齿状的力信号,这证明索烃分子间的氢键作用在受力解离之后仍可以恢复至原来的结合状态,体现出超分子相互作用的可逆性.通过对不同拉伸/松弛速率下的断裂(或成键)作用力进行统计分析,绘制出了该索烃体系解离及形成的能量路径.该聚合物交联后制备的功能化水凝胶的宏观力学性质,体现出了与单分子力学性质的直接关联(图18(a)).Fig.18(a)SchematicdescriptionoftheAFM-basedSMFSexperimentonpoly[2]catenaneandthepropertiesofcorrespondinghydrogels(ReproducedwithpermissionfromRef.[97] Copyright(2019)ChineseChemicalSociety) (b)Nanomechanicalpropertiesofasupramolecularhelix.Highdynamicflexibilityobtainedduringstretching(top)andlowstaticflexibilityasobtainedbythermalshape-fluctuationanalysis(bottom)(ReprintedwithpermissionfromRef.[100] Copyright(2020)Wiley-VCHVerlagGmbH&Co.KGaA,Weinheim).轮烷分子独特的超分子互锁结构使其成为了分子开关和分子马达等相关研究的模型分子.AFM单分子力谱被成功用于研究开启该分子马达所需要的外力大小以及动态解离与结合过程[98].实验过程中,轮烷分子的“轴”被固定在基底上,轮状分子则通过PEO桥联在AFM探针上.结果显示,轮烷分子主要依靠氢键来维持其结构稳定.在运动过程中,能量在轮状分子内氢键与PEO链和AFM微悬臂构成的串联体系的弹性势能间转换.研究人员又设计合成了通过机械互锁结构间相互作用形成类似蛋白质折叠结构的寡聚轮烷分子[98].在拉伸操纵寡聚轮烷分子的过程中,人们发现寡聚轮烷分子具有类似蛋白质的折叠性能,并且其折叠速率可以与蛋白质折叠速度相媲美.类似体系还包括基于多重氢键的UPy二聚体的力学蛋白模拟体系[99].此外AFM单分子力谱方法还被成功地用于由纯的超分子相互作用(π-π及疏水效应)所形成的超分子螺旋的纳米力学性质研究,研究发现该螺旋具有很低的静态柔性及很高的动态柔性(图18(b))[100].3.3生物大分子的纳米力学性质及动态结构研究3.3.1脱氧核糖核酸(DNA)DNA是生物体内重要的遗传物质,携带有转录RNA及合成蛋白质的重要信息.双链DNA独特的双螺旋结构对遗传信息的稳定存在及准确传递至关重要.在一些生理活动中,例如复制或转录等,双螺旋DNA经常受到外力作用发生一定程度的形变.因此,充分了解DNA的力学性质有助于深入理解一些重要的生理过程.近年来,随着DNA纳米技术的不断发展,各种基于DNA的纳米组装体被成功构筑,研究DNA互补链之间的结合强度对于功能组装体的构建及组装体的结构与性能调控具有重要意义[101].人们首先利用AFM单分子力谱研究了短双链DNA(10、20以及30个碱基对)的力学性质(图19(a))[30].结果显示,其断裂力值随着碱基数与力加载速率的增加而增加(图19(b)).此外,其断裂力值也受断裂方式的影响,即剪切断裂模式(2条链的5′端同时受力)所需的力值大于解拉链模式(2条链同一侧的5′端与3′端同时受力)[30].因此,短链核酸受力模式(剪切与解拉链)影响着其表观作用强度.Fig.19(a)Atypicalforce-piezodisplacementcurveofadsDNA (b)TheunbindingforcedependenceonthestretchingvelocityofdsDNA(10,20and30bp)(ReprintedwithpermissionfromRef.[30] Copyright(1999)NationalAcademyofSciences,U.S.A.).基于上述规律,研究人员将短双链DNA用作力学传感器[102].不同碱基对数量的短双链DNA被分别固定在PDMS模板和玻璃基底上,通过一段65碱基的带有荧光基团的单链DNA桥联.当模板与玻璃基底分离时,带有荧光基团的单链DNA会保留在碱基对数量较多的双链DNA一侧.该设计可以在碱基对数目相同的DNA中精确地区分出少至一个碱基对的错配.基于短双链DNA的力学稳定性,研究人员进一步将AFM单分子力谱与荧光显微镜结合,在单分子水平对短链DNA进行纳米组装[103].待搬运DNA的一端首先与存储区域基片表面固定的单股DNA形成双链结构,另一端则与针尖上固定的单链DNA互补形成桥联结构,针尖垂直运动过程中待搬运DNA以解拉链方式从基片表面断裂并留在针尖表面,从而被针尖从存储区域搬走.当该探针转移至目标样品区域时,被搬运DNA能够与该处基底固定的单股DNA形成一段新的双链DNA.此时,被搬运DNA与基片表面单链DNA形成的互补结构的长度大于其与针尖上的DNA形成的互补链的长度.当探针远离该区域时,串联结构中的两段双链DNA均处于剪切受力模式.由于被搬运DNA与目标区域基底固定的DNA形成的双链片段更长,稳定性更强,因此被搬运DNA与探针表面DNA解链,留在目标区域.利用上述原理,核酸分子可被连续搬运组装几百次,再结合荧光分子、量子点、蛋白质,可以实现多种物质在基片表面的图案化组装.通过将长链dsDNA桥联于AFM针尖及固体基片之间进行拉伸操纵,研究人员获得了具有指纹特征的长链dsDNA单分子力谱[9,104].在拉伸初期,DNA由无规卷曲变为伸展状态,对应力曲线的低力值区域.随着外力增加,DNA达到完全伸展长度后被进一步拉伸时,其构象由天然B-DNA变为过度拉伸的OS-DNA,导致力曲线出现高度约为65pN(F1)的长平台(B-OS转变,图20(a)).上述平台力值没有拉伸速率依赖性,具有指纹图谱特性.往复拉伸实验中,施加的外力等于或低于65pN时,松弛曲线可以与拉伸曲线重合.因此,B-OS转变是一个可逆过程.B-OS转变后继续拉伸,DNA将熔融解链,对应着力曲线高力值处的短平台(F2,图20(a)).该短平台力值受拉伸速率影响,属于非平衡过程.Fig.20(a)Atypicalforce-extensioncurveobtainedduringthestretchingofadsDNA(ReprintedwithpermissionfromRef.[9] Copyright(2012)EditorialOfficeofChemicalJournalofChineseUniversities) (b)AprobablemodeloftelomericDNAtopologyafterforce-inducedmelting.DifferentconditionsintherenaturationprocesseventuallyinducetelomericdsDNAtoformdifferenttopologies(ReprintedwithpermissionfromRef.[16] Copyright(2020)OxfordUniversityPress).长双链DNA的力学指纹谱可被用于探究长双链端粒序列在外力诱导下解链,及动态形成G-四链体(G4,由G链形成)及i-motif的过程(C4,由与G链互补的C链形成)[16].首先,研究人员开发了EM-PCR方法,合成了具有串联重复序列的长双链端粒DNA.随后,通过两段双链DNA把手序列以及金/巯基、生物素/链霉亲和素相互作用,将端粒DNA桥联于AFM探针与金基底之间.双链端粒DNA受力解链后,G-四链体和C-四链体的形成与双链DNA的复性相互竞争.单价K+、酸性条件、稳定i-motif结构或G-四链体结构的配体(如SWCNT和TMPyP4)均能够将平衡向抑制双链复性、促进高级结构形成的方向移动.只有当G-四链体和C-四链体同时形成时,双链结构的复性才会被完全抑制(图20(b)).DNA指纹图谱还能揭示小分子结合试剂对其力学稳定性的影响[9].例如:DNA小沟结合试剂纺锤菌素可以增加DNA的结构稳定性,表现为B-OS转变平台力值的增高,而另一种小沟结合剂偏端霉素A则导致平台力值降低.DNA大沟结合试剂α-helical肽及310-helical肽则使B-OS转变平台与DNA熔融解链所对应的平台融为一体.同时,高浓度的嵌入剂,例如溴化乙锭、道诺霉素、YO和普罗黄素等,使DNA链伸展,双螺旋结构消失,双链的力学稳定性增加,阻止外力诱导下的熔融解链,表现为B-OS转变平台的消失.共价交联剂顺式-二氨基二氯化铂通过交联DNA链中鸟嘌呤也能阻止外力诱导的熔融解链.上述结果表明,利用DNA指纹谱来探索DNA与小分子物质相互作用的研究,从单分子水平提供了小分子药物在体内的作用机理,为药物的筛选及研发提供了理论依据.长双链DNA的力学指纹谱还可用作DNA与DNA结合试剂(如DNA结合蛋白)相互作用的“指示剂”(图21).例如:上述DNA的B-OS转变过程,一直争议不断.一种观点认为,该过程中DNA的空间结构被破坏,双螺旋结构转变为梯形结构,但保持双链.另一种观点则认为,DNA发生了熔融解链.人们通过长双链DNA的指纹图谱及其与单链DNA结合蛋白(SSB)的相互作用验证上述2种观点[105].拉伸操纵DNA的过程中,SSB蛋白被加入(图21(a)).结果显示,该条件下的往复拉伸/松弛力曲线不能重合,出现了明显的滞后现象(图21(b)).表明B-OS转变过程中,部分双链DNA出现了熔融解链,并与SSB蛋白结合;松弛过程中,SSB蛋白阻碍单链DNA复性,表现为松弛曲线的滞后现象.根据滞后程度可以判断单股DNA结合试剂与其结合的程度及强度.Fig.21(a)SchematicillustrationofB-OStransitionofdsDNAandSSB-ssDNAinteraction (b)Typicalforce-extensioncurvesobtainedduringtherepeatedlymanipulatingofadsDNAinthepresenceofSSBproteins(ReprintedwithpermissionfromRef.[105] Copyright(2010)AmericanChemicalSociety) (c)SchematicillustrationoftheeffectofDnaDproteinondsDNA (d)Typicalforce-extensioncurvesobtainedbystretchingthesamedsDNAinthepresenceofDnaDprotein(ReprintedwithpermissionfromRef.[21] Copyright(2008)ElsevierLtd.).类似的方法还被用于探索双链DNA结合蛋白(例如DnaD蛋白)与双链DNA的相互作用[21].较高浓度的DnaD蛋白可以使双链DNA环化,形成圆盘样结构(图21(c)).因此,在该蛋白存在下拉伸双链DNA,力曲线上首先出现锯齿样力信号,对应圆盘结构被破坏的过程(图21(d)).此后,该蛋白会促进双链DNA解螺旋,导致其完全伸展长度变长,表现为力曲线上对应B-OS转变平台的变短及右移(图21(d)).3.3.2多糖(船式-椅式转变)多聚糖是重要的能源物质,其糖环结构、连接方式及分子内氢键使其具有独特的单分子力学性质[1,9,29,106].多聚糖中连接吡喃糖环的键的取向与糖环长轴的相对关系决定了力曲线的形态.二者相互取向垂直时,会在力曲线上产生“肩式”平台,对应糖环船式-椅式结构的转变.这种糖环构象转变对应的“肩式”平台高度恒定,具有指纹图谱特征,可以用于共价键等强相互作用体系研究的单分子信号指示剂.例如:人们曾用羧甲基化淀粉作间隔基团,以其构象转变的指纹图谱作“指示剂”,测量了单个Au―S(Au―Au)键、Si―C键、组氨酸与Ni2+及NTA之间的络合作用的强度等等[40,107].有些多糖分子在溶液中能形成螺旋结构,被拉伸操纵时会获得带有长平台的力信号[2,108].以淀粉分子为例,拉伸操纵的初期,淀粉链由无规卷曲变为伸展状态,对应力曲线上力值接近基线的部分;随后,淀粉链内氢键断裂,空间结构解螺旋,出现长平台样力信号;外力逐渐增大,淀粉链内糖环的构象由椅式转变为船式,高力值区域出现“肩式”平台(图22)[2,108].Fig.22Force-extensioncurvesobtainedduringthestretchingofasingleamylosechaininbutanol(ReprintedwithpermissionfromRef.[108] Copyright(2006)AmericanChemicalSociety).3.3.3蛋白质蛋白质的空间结构对其性质与功能有重要影响.单分子力谱是研究蛋白质折叠结构及动态解折叠与折叠过程的有效手段[9,29,109,110].相关研究始于对机械力极其敏感的肌联蛋白作为研究对象[110,111].多聚肌联蛋白被固定在探针与基底之间,在探针远离基底的过程中,类似免疫球蛋白的结构域在外力作用下依次解折叠,产生了锯齿形力信号.锯齿峰间的距离约为25~28nm,这与解折叠结构域的完全伸展长度(31nm)非常近似.因此,锯齿峰对应着肌联蛋白中免疫球蛋白样结构域的解折叠过程.此后,在拉伸具有确定数目的免疫球蛋白样串联结构域(Ig4或Ig8)时,得到了最多含有4或8个连续锯齿峰的力信号,再次验证锯齿峰对应着结构域的解折叠.此外,拉伸后松弛,蛋白质可以再次折叠.这也证明,外力诱导下,一些蛋白质的折叠及解折叠过程是可逆的.同时,蛋白质的解折叠指纹谱可以作为“指示剂”,探究自身的错误折叠及其他蛋白质的折叠与解折叠过程[109,112,113].蛋白质的力学指纹谱还能用于研究蛋白质与蛋白质或其他物质的相互作用,直接探索参与相互作用的结构域等.例如:人们将重组蛋白(NuG2)8固定在探针与基底之间,在拉伸过程中,加入相互作用的hFc蛋白[114].结果显示(NuG2)8解折叠力曲线上锯齿峰的力值显著增加.这证明hFc的结合增加了(NuG2)8结构的稳定性.利用AFM单分子力谱获得了蛋白质的微观力学性质后,人员尝试利用这些信息预测宏观材料的力学性能并指导新型材料的设计[17].例如:GB1蛋白在解折叠后能够快速地重新折叠,节肢弹性蛋白(resilin,R)能表现出显著的熵弹性.因此,研究人员预测上述2种蛋白组成的融合蛋白将展现良好的韧性与弹性.通过将GB1蛋白与节肢弹性蛋白进行交联,得到了水凝胶样的宏观材料.低应变条件下,该材料在节肢弹性蛋白的贡献下展现出很好的形变恢复能力;而在较高应变情况下,GB1的存在使该材料具有能量耗散性能,显著提高了材料的力学性能.该研究有效建立了生物大分子微观力学性能与材料宏观力学性能之间的联系.3.3.4真实生物体系-天然病毒从单个分子水平探测活体细胞中生物大分子间的相互作用对于理解这些大分子参与的生命过程的机制,继而实现对相关过程的调控具有重要意义,然而相关研究充满挑战.病毒是介于生物大分子与活体细胞的一个中间状态,其结构相对简单.研究人员选取烟草花叶病毒为模型体系,利用AFM单分子力谱探测了该病毒中RNA与蛋白质外壳的相互作用及解组装过程(图23)[115].首先利用金-巯基相互作用将病毒颗粒“垂直”地固定在金基底.当探针与病毒颗粒接触时,病毒RNA通过物理作用吸附在探针上.当AFM探针远离基底时,RNA在探针的牵引下从蛋白质外壳上解组装(图23(a)与图23(b)).上述过程获得了带有锯齿的平台状力信号,平台的力值大小依赖于RNA的拉伸速率,对应着RNA与蛋白质外壳间相互作用的强度.此外,力曲线上相邻2个锯齿峰间的距离与病毒RNA中“AAG”序列(与蛋白质外壳作用最强的序列)间的距离相似,表明“锯齿”状力信号对应不同序列的RNA从蛋白质外壳上的解组装(图23(c)).在此基础上,通过系统研究溶液pH值、钙离子浓度等对RNA解组装过程的影响规律,揭示了该病毒从外界进入植物细胞后边解组装边复制的分子机理[51].Fig.23(a)TMVStructure (b)SchematicillustrationoftheRNAmanipulationonTMV (c)Comparisonofthedistancebetweenadjacentsawtooth-likepeaksinstretchingcurvesandtheoreticaldistancebetweenadjacentAAGsintheTMVRNAsequence(ReprintedwithpermissionfromRef.[115] Copyright(2010)AmericanChemicalSociety) (d)Force-extensioncurvesobtainedduringthepullingofRNAoutofTMVintheabsence(blue)andpresence(red)oftannin(ReprintedwithpermissionfromRef.[116] Copyright(2019)TheRoyalSocietyofChemistry).随后,研究人员探究了单宁酸及其衍生物对烟草花叶病毒RNA与其蛋白质外壳相互作用的影响,揭示抗病毒药物的作用机理[116].结果显示,加入单宁酸后将RNA从TMV病毒上解组装所需外力显著增大(图23(d)),即单宁酸能显著增加病毒RNA(尤其是AAG序列)与蛋白质外壳的相互作用,阻碍病毒在生物体内的解组装.这是由于单宁酸上的棓酰基与蛋白质、核酸的活性基团之间形成了较多非共价键,例如疏水相互作用和氢键等.同时,当RNA被拉伸出病毒颗粒后,单宁酸迅速占据蛋白外壳内的RNA结合位点,导致RNA无法有序地重新组装.该项工作从单分子水平深化了人们对单宁酸的抗病毒机制的认识,有助于指导设计新型的抗病毒药物.4结语及展望随着AFM技术及相关仪器的不断进步,以及相关实验经验的积累,AFM单分子力谱的适用对象已经从简单的孤立大分子体系拓展到复杂、真实体系[13,115,116].同时,为了降低流体力学阻力,在接近材料真实使用环境中研究高分子链的行为,AFM单分子力谱的探测微环境从液相被拓展到空气相[117].通过优化环境湿度、探针的弹性系数及疏水性来克服黏附力影响,研究人员成功地在空气相中探索了PEO单晶的力致熔融机制.为减小环境介质对大分子本征性质的干扰,AFM单分子力谱的工作微环境又被拓展到真空,并精确测量了PS分子中π-π作用[57,63].为了观察一些快速的动力学过程及捕捉存在时间极短的中间态,AFM探针被进一步优化,具有了更快的响应速度.微悬臂的探测速率与其共振频率相关,通过减小微悬臂的尺寸(尤其是长度)可以增加其弹性系数,进而增加共振频率,由于流体力学阻力也随之减小,该探针的探测速率最终被提高至3870µm/s[34].利用该微型探针,人们获得了肌肉纤维中肌联蛋白(Titin)在高速拉伸时的解折叠信息,结合分子动力学模拟(通常在更高的拉伸速率下进行),有助于深入理解其折叠与解折叠机制.此外,研究人员还利用聚焦离子束改变微悬臂的形状与尺寸,通过降低其流体力学阻力与硬度来提高力精度与力稳定性[35].加工后,微悬臂的共振频率几乎没有变化,但流体力学系数及弹性系数显著降低,噪音被控制在sub-pN范围.AFM技术与其他光谱技术结合,在获得力学及长度信息的同时还能获得化学组成信息,将有助于更好地建立材料结构与性能之间的联系.例如:基于AFM探针的针尖增强拉曼光谱(TERS),可以同时获得材料的表面形貌及表面物种的光谱信息[118];AFM与红外光谱联用的纳米红外系统,能够将样品的形貌、性能和化学结构等信息很好地统一(图24)[119].同样,AFM单分子力谱技术与其他光谱技术的原位结合将使得人们对于单分子的力学、光学及电学等的原位研究成为可能,将为功能材料的开发奠定重要基础.Fig.24(a)SchematicdiagramofAFM-IR (b)TheAFMprobeoscillationinresponsetoIRabsorptionofthesample (c)ThemeasurementoftheAFMprobeoscillationasafunctionofwavenumber(ReprintedwithpermissionfromRef.[119] Copyright(2017)AmericanChemicalSociety).参考文献1ZhangWK,ZhangX.ProgPolymSci,2003,28(8):1271-1295.doi:10.1016/s0079-6700(03)00046-72ZhangX,LiuCJ,WangZQ.Polymer,2008,49(16):3353-3361.doi:10.1016/j.polymer.2008.04.0563NoyA.CurrOpinChemBiol,2011,15(5):710-718.doi:10.1016/j.cbpa.2011.07.0204JanshoffA,NeitzertM,OberdörferY,FuchsH.AngewChemIntEd,2000,39(18):3212-3237.doi:3.0.co 2-x%22\t%22http://www.gfzxb.org/thesisDetails%22\l%2210.11777/_blank"10.1002/1521-3773(20000915)39:183.0.co 2-x5ButtHJ,CappellaB,KapplM.SurfSciRep,2005,59(1-6):1-152.doi:10.1016/j.surfrep.2005.08.0036GunariN,BalazsAC,WalkerGC.JAmChemSoc,2007,129(33):10046-10047.doi:10.1021/ja068652w7MarszalekPE,LiH,OberhauserAF,FernandezJM.ProcNatlAcadSciUSA,2002,99(7):4278-4283.doi:10.1073/pnas.0724356998PangXX,CuiSX.Langmuir,2013,29(39):12176-12182.doi:10.1021/la403132e9ZhangW,KouXL,ZhangWK.ChemJChineseU,2012,33(5):861-87510XiangWT,LiZD,XuCQ,LiJ,ZhangWK,XuHP.ChemAsianJ,2019,14(9):1481-1486.doi:10.1002/asia.20190033211ZhangH,LiX,LinYG,GaoF,TangZ,SuPF,ZhangWK,XuYZ,WengWG,BoulatovR.NatCommun,2017,8:1147.doi:10.1038/s41467-017-01412-812RajG,BalnoisE,BaleyC,GrohensY.JScannProbeMicrosc,2009,4(2):1-7.doi:10.1166/jspm.2009.101013LiuK,SongY,FengW,LiuNN,ZhangWK,ZhangX.JAmChemSoc,2011,133(10):3226-3229.doi:10.1021/ja108022h14SpruijtE,vandenBergSA,CohenStuartMA,vanderGuchtJ.ACSNano,2012,6(6):5297-5303.doi:10.1021/nn301097y15EubelenM,BostailleN,CabochetteP,GauquierA,TebabiP,DumitruAC,KoehlerM,GutP,AlsteensD,StainierDYR,Garcia-PinoA,VanhollebekeB.Science,2018,361(6403):eaat1178.doi:10.1126/science.aat117816ZhangXN,ZhangYQ,ZhangWK.NucleicAcidsRes,2020,48(12):6458-6470.doi:10.1093/nar/gkaa47917LvSS,DudekDM,CaoY,BalamuraliMM,GoslineJ,LiHB.Nature,2010,465(7294):69-73.doi:10.1038/nature0902418SchonfelderJ,Perez-JimenezR,MunozV.NatCommun,2016,7:11777.doi:10.1038/ncomms1177719LiuNN,ZhangWK.ChemPhysChem,2012,13(9):2238-2256.doi:10.1002/cphc.20120015420ZhangW,LuXJ,ZhangWK,ShenJC.Langmuir,2011,27(24):15008-15015.doi:10.1021/la203752y21ZhangWK,MachonC,OrtaA,PhillipsN,RobertsCJ,AllenS,SoultanasP.JMolBiol,2008,377(3):706-714.doi:10.1016/j.jmb.2008.01.06722LiuJY,FengW,ZhangWK.Nanoscale,2020,12(6):4159-4166.doi:10.1039/c9nr09054a23ZlatanovaJ,LeubaSH.JMuscleResCellMotil,2002,23(5-6):377-395.doi:10.1023/a:102349812045824ZlatanovaJ,LeubaSH.JMolBiol,2003,331(1):1-19.doi:10.1016/s0022-2836(03)00691-025ZhangWenke(张文科).ActaPolymencaSinica(高分子学报),2011,(9):913-922.doi:10.3979/j.issn.1673-825X.2011.04.00726KimY,KimW,ParkJW.BullKoreanChemSoc,2016,37(12):1895-1907.doi:10.1002/bkcs.1102227FriedsamC,GaubHE,NetzRR.Biointerphases,2006,1(1):MR1-MR21.doi:10.1116/1.217199628LiuY,JuliusVancsoG.ProgPolymSci,2020,104:101232.doi:10.1016/j.progpolymsci.2020.10123229ZhangWK,WangC,ZhangX.ChineseSciBull,2003,48(11):1113-1126.doi:10.1360/02wd040930StrunzT,OroszlanK,SchaferR,GuntherodtHJ.ProcNatlAcadSciUSA,1999,96(20):11277-11282.doi:10.1073/pnas.96.20.1127731HuangW,WuX,GaoX,YuY,LeiH,ZhuZ,ShiY,ChenY,QinM,WangW,CaoY.NatChem,2019,11(4):310-319.doi:10.1038/s41557-018-0209-232LyuXJ,SongY,FengW,ZhangWK.ACSMacroLett,2018,7(6):762-766.doi:10.1021/acsmacrolett.8b0035533EdwardsDT,PerkinsTT.JStructBiol,2017,197(1):13-25.doi:10.1016/j.jsb.2016.01.00934RicoF,GonzalezL,CasusoI,Puig-VidalM,ScheuringS.Science,2013,342(6159):741-753.doi:10.1126/science.123976435FaulkJK,EdwardsDT,BullMS,PerkinsTT.MethodsEnzymol,2017,582:321-351.doi:10.1016/bs.mie.2016.08.00736YuH,SiewnyMGW,EdwardsDT,SandersAW,PerkinsTT.Science,2017,355(6328):945-950.doi:10.1126/science.aah712437LiHB,RiefM,OesterheltF,GaubHE.AdvMater,1998,10(4):316-319.doi:3.0.co 2-a%22\t%22http://www.gfzxb.org/thesisDetails%22\l%2210.11777/_blank"10.1002/(sici)1521-4095(199803)10:43.0.co 2-a38MarszalekPE,OberhauserAF,PangYP,FernandezJM.Nature,1998,396(6712):661-664.doi:10.1038/2532239RiefM,OesterheltF,HeymannB,GaubHE.Science,1997,275(5304):1295.doi:10.1126/science.275.5304.129540GrandboisM,BeyerM,RiefM,Clausen-SchaumannH,GaubHE.Science,1999,283(5408):1727.doi:10.1126/science.283.5408.172741LiHB,RiefM,OesterheltF,GaubHE,ZhangX,ShenJC.ChemPhysLett,1999,305(3-4):197-201.doi:10.1016/s0009-2614(99)00389-942ZhangB,ShiR,DuanW,LuoZ,LuZY,CuiS.RSCAdv,2017,7(54):33883-33889.doi:10.1039/c7ra05779b43FlorinEL,MoyVT,GaubHE.Science,1994,264(5157):415-417.doi:10.1126/science.815362844SikoraAE,SmithJR,CampbellSA,FirmanK.SoftMatter,2012,8(23):6358-6363.doi:10.1039/c2sm07213k45HinterdorferP,DufreneYF.NatMethods,2006,3(5):347-355.doi:10.1038/nmeth87146XueYR,LiX,LiHB,ZhangWK.NatCommun,2014,5:4348.doi:10.1038/ncomms534847ZhangWK.Nano-mechanicalDetectionofSingleMolecules.DoctoralDissertationofJilinUniversity,200248DengYB,WuT,WangMD,ShiSC,YuanGD,LiX,ChongHC,WuB,ZhengP.NatCommun,2019,10:2775.doi:10.1038/s41467-019-10696-x49JohnsonKC,ThomasWE.BiophysJ,2018,114(9):2032-2039.doi:10.1016/j.bpj.2018.04.00250LiZD,SongY,LiAS,XuWQ,ZhangWK.Nanoscale,2018,10(39):18586-18596.doi:10.1039/c8nr06150e51LiuNN,ChenY,PengB,LinY,WangQ,SuZH,ZhangWK,LiHB,ShenJC.BiophysJ,2013,105(12):2790-2800.doi:10.1016/j.bpj.2013.10.00552CaoY,LiHB.Langmuir,2011,27(4):1440-1447.doi:10.1021/la104130n53PopaI,KosuriP,Alegre-CebolladaJ,Garcia-ManyesS,FernandezJM.NatProtoc,2013,8(7):1261-1276.doi:10.1038/nprot.2013.05654OberhauserAF,HansmaPK,Carrion-VazquezM,FernandezJM.ProcNatlAcadSciUSA,2001,98(2):468-472.doi:10.1073/pnas.98.2.46855SulchekT,FriddleRW,NoyA.BiophysJ,2006,90(12):4686-4691.doi:10.1529/biophysj.105.08029156WeiHZ,vandeVenTGM.ApplSpectroscRev,2008,43(2):111-133.doi:10.1080/0570492070183125457BaoY,LuoZL,CuiSX.ChemSocRev,2020,49(9):2799-2827.doi:10.1039/c9cs00855a58YuY,ZhangYH,JiangZH,ZhangX,ZhangHM,WangXH.Langmuir,2009,25(17):10002-10006.doi:10.1021/la901169p59ZhangWK,ZouS,WangC,ZhangX.JPhysChemB,2000,104(44):10258-10264.doi:10.1021/jp000459f60ShiWQ,ZhangYH,LiuCJ,WangZQ,ZhangX,ZhangYH,ChenYM.Polymer,2006,47(7):2499-2504.doi:10.1016/j.polymer.2005.12.08961LiIT,WalkerGC.AccChemRes,2012,45(11):2011-2021.doi:10.1021/ar200285h62LiIT,WalkerGC.JAmChemSoc,2010,132(18):6530-6540.doi:10.1021/ja101155h63CaiWH,XuD,QianL,WeiJH,XiaoC,QianLM,LuZY,CuiSX.JAmChemSoc,2019,141(24):9500-9503.doi:10.1021/jacs.9b0349064DiWS,GaoX,HuangWM,SunY,LeiH,LiuY,LiWF,LiYR,WangX,QinM,ZhuZS,CaoY,WangW.PhysRevLett,2019,122(4):047801-047806.doi:10.1103/physrevlett.122.04780165ZhangS,QianHJ,LiuZH,JuHY,LuZY,ZhangHM,ChiLF,CuiSX.AngewChemIntEd,2019,58(6):1659-1663.doi:10.1002/anie.20181115266DelparastanP,MalollariKG,LeeH,MessersmithPB.AngewChemIntEd,2019,58(4):1077-1082.doi:10.1002/anie.20181176367BalzerBN,GalleiM,HaufMV,StallhoferM,WieglebL,HolleitnerA,RehahnM,HugelT.AngewChemIntEd,2013,52(25):6541-6544.doi:10.1002/anie.20130125568FengW,WangZG,ZhangWK.Langmuir,2017,33(8):1826-1833.doi:10.1021/acs.langmuir.6b0445769SongY,MaZW,YangP,ZhangXY,LyuXJ,JiangK,ZhangWK.Macromolecules,2019,52(3):1327-1333.doi:10.1021/acs.macromol.8b0270270MaZW,YangP,ZhangXY,JiangK,SongY,ZhangWK.ACSMacroLett,2019,8(9):1194-1199.doi:10.1021/acsmacrolett.9b0060771LiJ,NagamaniC,MooreJS.AccChemRes,2015,48(8):2181-2190.doi:10.1021/acs.accounts.5b0018472CraigSL.Nature,2012,487(7406):176-177.doi:10.1038/487176a73ChenZX,MercerJAM,ZhuXL,RomaniukJAH,PfattnerR,CegelskiL,MartinezTJ,BurnsNZ,XiaY.Science,2017,357(6350):475-479.doi:10.1126/science.aan279774KeanZS,NiuZB,HewageGB,RheingoldAL,CraigSL.JAmChemSoc,2013,135(36):13598-13604.doi:10.1021/ja407599775GossweilerGR,KouznetsovaTB,CraigSL.JAmChemSoc,2015,137(19):6148-6151.doi:10.1021/jacs.5b0249276WangJP,KouznetsovaTB,NiuZB,OngMT,KlukovichHM,RheingoldAL,MartinezTJ,CraigSL.NatChem,2015,7(4):323-327.doi:10.1038/nchem.218577DavisDA,HamiltonA,YangJL,CremarLD,vanGoughD,PotisekSL,OngMT,BraunPV,MartinezTJ,WhiteSR,MooreJS,SottosNR.Nature,2009,459(7243):68-72.doi:10.1038/nature0797078HickenbothCR,MooreJS,WhiteSR,SottosNR,BaudryJ,WilsonSR.Nature,2007,446(7134):423-427.doi:10.1038/nature0568179WangJP,KouznetsovaTB,CraigSL.JAmChemSoc,2015,137(36):11554-11557.doi:10.1021/jacs.5b0616880RobbMJ,KimTA,HalmesAJ,WhiteSR,SottosNR,MooreJS.JAmChemSoc,2016,138(38):12328-12331.doi:10.1021/jacs.6b0761081KouznetsovaTB,WangJP,CraigSL.ChemPhysChem,2017,18(11):1486-1489.doi:10.1002/cphc.20160046382KlukovichHM,KeanZS,BlackRamirezAL,LenhardtJM,LinJX,HuXQ,CraigSL.JAmChemSoc,2012,134(23):9577-9580.doi:10.1021/ja302996n83KlukovichHM,KeanZS,IaconoST,CraigSL.JAmChemSoc,2011,133(44):17882-17888.doi:10.1021/ja207451784KeanZS,BlackRamirezAL,YanYF,CraigSL.JAmChemSoc,2012,134(31):12939-12942.doi:10.1021/ja306366685KrygerMJ,MunarettoAM,MooreJS.JAmChemSoc,2011,133(46):18992-18998.doi:10.1021/ja208672886WangJP,OngMT,KouznetsovaTB,LenhardtJM,MartinezTJ,CraigSL.JOrgChem,2015,80(23):11773-11778.doi:10.1021/acs.joc.5b0149387CarusoMM,DavisDA,ShenQ,OdomSA,SottosNR,WhiteSR,MooreJS.ChemRev,2009,109(11):5755-5798.doi:10.1021/cr900135388PotisekSL,DavisDA,SottosNR,WhiteSR,MooreJS.JAmChemSoc,2007,129(45):13808-13809.doi:10.1021/ja076189x89WuD,LenhardtJM,BlackAL,AkhremitchevBB,CraigSL.JAmChemSoc,2010,132(45):15936-15938.doi:10.1021/ja108429h90SerpeMJ,KerseyFR,WhiteheadJR,WilsonSM,ClarkRL,CraigSL.JPhysChemC,2008,112(49):19163-19167.doi:10.1021/jp806649a91LenhardtJM,BlackAL,CraigSL.JAmChemSoc,2009,131(31):10818-10819.doi:10.1021/ja903654892PotisekSL,DavisDA,SottosNR,WhiteSR,MooreJS.JAmChemSoc,2007,129(45):13808-13809.doi:10.1021/ja076189x93ChengB,CuiSX.TopCurrChem,2015,369:97-134.doi:10.1007/128_2015_62894LiuYL,WangZQ,ZhangX.ChemSocRev,2012,41(18):5922-5932.doi:10.1039/c2cs35084j95ZouS,SchönherrH,VancsoGJ.AngewChemIntEd,2005,44(6):956-959.doi:10.1002/anie.20046096396LiuYL,YuY,GaoJ,WangZQ,ZhangX.AngewChemIntEd,2010,49(37):6576-6579.doi:10.1002/anie.20100241597XingH,LiZD,WangWB,LiuPR,LiuJK,SongY,WuZL,ZhangWK,HuangFH.CCSChem,2019,1:513-523.doi:10.31635/ccschem.019.2019004398SluysmansD,HubertS,BrunsCJ,ZhuZX,StoddartJF,DuwezAS.NatNanotechnol,2018,13(3):209-213.doi:10.1038/s41565-017-0033-799ChungJ,KushnerAM,WeismanAC,GuanZ.NatMater,2014,13(11):1055-1062.doi:10.1038/nmat4090100WangHJ,ShenBW,SongY,LeeMS,ZhangWK.MacromolRapidCommun,2020,41(24):2000453.doi:10.1002/marc.202000453101RothemundPWK.Nature,2006,440(7082):297-302.doi:10.1038/nature04586102AlbrechtC,BlankK,Lalic-MülthalerM,HirlerS,MaiT,GilbertI,SchiffmannS,BayerT,Clausen-SchaumannH,GaubHE.Science,2003,301(5631):367-370.doi:10.1126/science.1084713103KuferSK,PuchnerEM,GumppH,LiedlT,GaubHE.Science,2008,319(5863):594-596.doi:10.1126/science.1151424104RiefM,Clausen-SchaumannH,GaubHE.NatStructBiol,1999,6(4):346-349.doi:10.1038/7582105LiuNN,BuTJ,SongY,ZhangW,LiJJ,ZhangWK,ShenJC,LiHB.Langmuir,2010,26(12):9491-9496.doi:10.1021/la100037z106MarszalekPE,PangYP,LiHB,YazalJE,OberhauserAF,FernandezJM.ProcNatlAcadSciUSA,1999,96(14):7894-7898.doi:10.1073/pnas.96.14.7894107ContiM,FaliniG,SamorìB.AngewChemIntEd,2000,39(1):215-218.doi:3.0.co 2-r%22\t%22http://www.gfzxb.org/thesisDetails%22\l%2210.11777/_blank"10.1002/(sici)1521-3773(20000103)39:13.0.co 2-r108ZhangQM,LuZY,HuH,YangWT,MarszalekPE.JAmChemSoc,2006,128(29):9387-9393.doi:10.1021/ja057693+109MarszalekPE,DufreneYF.ChemSocRev,2012,41(9):3523-3534.doi:10.1039/c2cs15329g110FisherTE,OberhauserAF,Carrion-VazquezM,MarszalekPE,FernandezJM.TrendsBiochemSci,1999,24(10):379-384.doi:10.1016/s0968-0004(99)01453-x111RiefM,GautelM,OesterheltF,FernandezJM,GaubHE.Science,1997,276(5315):1109-1112.doi:10.1126/science.276.5315.1109112OberhauserAF,MarszalekPE,Carrion-VazquezM,FernandezJM.NatStructBiol,1999,6(11):1025-1028.doi:10.1038/14907113DelRioA,Perez-JimenezR,LiuRC,Roca-CusachsP,FernandezJM,SheetzMP.Science,2009,323(5914):638-641.doi:10.1126/science.1162912114CaoY,BalamuraliMM,SharmaD,LiH.ProcNatlAcadSciUSA,2007,104(40):15677-15681.doi:10.1073/pnas.0705367104115LiuNN,PengB,LinY,SuZH,NiuZW,WangQ,ZhangWK,LiHB,ShenJC.JAmChemSoc,2010,132(32):11036-11038.doi:10.1021/ja1052544116WangHJ,ChenY,ZhangWK.Nanoscale,2019,11(35):16368-16376.doi:10.1039/c9nr05410c117YangP,SongY,FengW,ZhangWK.Macromolecules,2018,51(18):7052-7060.doi:10.1021/acs.macromol.8b01544118Deckert-GaudigT,TaguchiA,KawataS,DeckertV.ChemSocRev,2017,46(13):4077-4110.doi:10.1039/c7cs00209b119DazziA,PraterCB.ChemRev,2017,117(7):5146-5173.doi:10.1021/acs.chemrev.6b00448原文链接:http://www.gfzxb.org/thesisDetails#10.11777/j.issn1000-3304.2020.20266&lang=zhDOI:10.11777/j.issn1000-3304.2020.20266《高分子学报》高分子表征技术专题链接:http://www.gfzxb.org/article/doi/10.11777/j.issn1000-3304




















 我要推广仪器
我要推广仪器
 下载APP
下载APP