用[url=https://insevent.instrument.com.cn/t/Mp][color=#3333ff]气相色谱[/color][/url]建立乙酸含量的标准曲线,怎么配制标准品呀,FID检测器
大家好,对于气相色谱我以前一直用的是面积归一法,现在想尝试用外标法做产品的含量,可是标准曲线做出来了,而且和做标线一起分析的未知样能够直接得到结果,而我重新进样后的峰面积什么的也都能出来,可是浓度栏确都是零,重新加载方法后还是没有结果。请大家帮我分析下可能是哪里出了问题。噢我用的是岛津的GC2010和GC2014的色谱。
用高效液相色谱仪外标法测定兽药含量可不可以不建立标准曲线?测出标准品和样品的色谱峰之后直接带入公式求含量可以吗?
一、标准曲线一般用分光光度法测物质的含量,先要制作标准曲线,然后根据标准曲线查出所测物质的含量。因此,制作标准曲线是生物检测分析的一项基本技术。二、蛋白质含量测定方法 1. 凯氏定氮法2. 双缩脲法3. Folin-酚试剂法4. 紫外吸收法5. 考马斯亮蓝法三、考马斯亮蓝法测定蛋白质含量—标准曲线制作 (一)试剂: 1. 考马斯亮蓝试剂:考马斯亮蓝G—250 100 mg溶于50 ml 95%乙醇,加入100 ml 85% H3 PO4 ,雍蒸馏水稀释至1000 ml,滤纸过滤。最终试剂中含0.01%(W/V)考马斯亮蓝G—250,4.7%(W/V)乙醇,8.5%(W/V)H3 PO4 。2. 标准蛋白质溶液:纯的牛血清血蛋白,预先经微量凯氏定氮法测定蛋白氮含量,根据其纯度同0.15 mol/LNaCl配制成100 ug/ml蛋白溶液。(二)器材: 1. 722S型分光光度计使用及原理。2. 移液管使用。(三)标准曲线制作: 1. 试管编号0123456100ug/ml标准蛋白(ml)0.00.10.20.30.40.50.60.15mol/L NaCl (ml)10.90.80.70.60.50.4考马斯亮蓝试剂 (ml)5555555摇匀,1h内以1号管为空白对照,在595nm处比色A595nm2. 以A595nm 为纵坐标,标准蛋白含量为横坐标(六个点为10ug、20 ug、30 ug、40 ug、50 ug、60 ug),在坐标轴上绘制标准曲线。(1)利用标准曲线查出回归方程。(2)用公式计算回归方程。(3)或用origin作图 ,测出回归线性方程。即A595nm =a×X( )+6;一般相关系数应过0.999以上,至少2个9以上。(4)绘图时近两使点在一条直线上,在直线上的点应该在直线两侧。(四)蛋白质含量的测定: 样品即所测蛋白质含量样品(含量应处理在所测范围内),依照操作步骤1操作,测出样品的A595nm ,然后利用标准曲线或回归方程求出样品蛋白质含量。一般被测样品的A595nm 值在0.1—0.05之间,所以上述样品如果A595nm 值太大,可以稀释后再测A595nm值,然后再计算。(五)注意事项: 1. 玻璃仪器要洗涤干净。2. 取量要准确。3. 玻璃仪器要干燥,避免温度变化。4. 对照:用被测物质以外的物质作空白对照。蛋白质含量测定实验难度系数 0共 0 人点评打分实验材料结晶牛血清清蛋白 g—球蛋白试剂(盒)Na2CO3酒石酸钾钠蒸馏水硫酸铜钨酸钠钼酸钠磷酸硫酸锂液体溴NaOH酚酞仪器耗材可见光分光光度计旋涡混合器秒表[url=http://www.biomart.cn/equipm
原吸标准液浓度我使用AAS—400测定待测溶液中元素含量,使用的是标准曲线法,所取得标准液浓度为0.2 0.4 0.6 0.8 1.0g/l我觉得是不是标准溶液的浓度太大了,做出来的工作曲线不是直线。待测溶液的原子浓度很低,只有零点几毫克每升
用液相标准曲线法检测含量时,如果样品面积在标准曲线范围之内,但接近上限,对含量计算有影响?
大家好 请教个问题 我做液相色谱分析标准曲线,用邻氯苯胺盐酸盐做标准曲线,但是样品是邻氯苯胺硫酸盐,怎么计算邻氯苯胺的含量?谢谢
求助,使用气相色谱法测定空气中正己烷,正庚烷,正戊烷的含量,因为客观条件所限不能购买标准气,使用自配的标准气准确度太差。想问如果用标准溶液建一条标准曲线,可不可以将液体中的浓度通过某种方法换算为气体中的浓度,然后进行气体中同种物质的定量检测?或者说不用通过换算可以直接使用标准溶液建立的标准曲线呢?
[color=#444444]使用[url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]法测定空气中正己烷,正庚烷,正戊烷的含量,因为客观条件所限不能购买标准气,使用自配的标准气准确度太差。想问如果用标准溶液建一条标准曲线,可不可以将液体中的浓度通过某种方法换算为气体中的浓度,然后进行气体中同种物质的定量检测?或者说不用通过换算可以直接使用标准溶液建立的标准曲线呢?[/color]
急!急!急!麻烦懂行的达达们帮我设计个气相色谱外标法标准曲线的操作流程。是做板材中TVOC的含量的,标准样品有4针3个不同浓度,一针是空白。请问标准样品需要稀释到一定浓度还是可以直接进样。还有怎么做标准曲线啊?
求标准《SN/T 2380-2009 石油产品中芳烃含量的测定 高效液相色谱法》,标准版面太冷清了,故在此发帖。谢谢!
求助标准SN/T 3727-2013 《进出口食品中碘含量的测定 离子色谱法》,谢谢!
在分析化学中,特别是仪器分析实验中,经常用某物质已校正的标准曲线来求相应物质的未知浓度。标准曲线通常是一条过原点的直线,被测组分含量可从标准曲线上求得。以分光光度计法为例,某特定波长的光经过某物质,可以被该物质吸收、反射等,并且其浓度(c)与吸光度(A)之间在一定浓度范围内符合朗伯-比尔定律。 但有时由于人为操作误差、仪器系统不稳定等因素,所有的实验点往往不在同一条直线上,这就需要用数理统计方法找出各数据点误差最小的直线,对数据进行用最小二乘法分析,求出回归方程,然后绘制回归曲线。标准曲线法和标准加入法是大家经常会搞混的方法。简而言之,标准曲线法适用于组成较为简单的大批量式样测定,操作比较简单;标准加入法能有效消除集体不同造成的误差,提高分析结果的准确性,但工作量大。标准曲线法和标准加入法区别1、标准曲线法也称外标法或直接比较法,是一种简便、快速的定量方法。与分光光度分析中的标准曲线法相似,首先用欲测组分的标准样品绘制标准曲线。具体方法是:用标准样品配制成不同浓度的标准系列,在与待测组分相同的色谱条件下,等体积准确进样,测量各峰的峰面积或峰高,用峰面积或峰高对样品浓度绘制标准曲线,此标准曲线应是通过原点的直线。若标准曲线不通过原点,则说明存在系统误差。标准曲线的斜率即为绝对校正因子。在测定样品中的组分含量时,要用与绘制标准曲线完全相同的色谱条件作出色谱图,测量色谱峰面积或峰高,然后根据峰面积和峰高在标准曲线上直接查出注入色谱柱中样品组分的浓度。标准曲线法的优点是:绘制好标准工作曲线后测定工作就变得相当简单,可直接从标准工作曲线上读出含量,因此特别适合于大量样品的分析。标准曲线法的缺点是:每次样品分析的色谱条件(检测器的响应性能,柱温,流动相流速及组成,进样量,柱效等)很难完全相同,因此容易出现较大误差。此外,标准工作曲线绘制时,一般使用欲测组分的标准样品(或已知准确含量的样品),而实际样品的组成却千差万别,因此必将给测量带来一定的误差。2、标准加入法是一种被广泛使用的检验仪器准确度的测试方法。这种方法尤其适用于检验样品中是否存在干扰物质。具体方法是:将一定量已知浓度的标准溶液加入待测样品中,测定加入前后样品的浓度。加入标准溶液后的浓度将比加入前的高,其增加的量应等于加入的标准溶液中所含的待测物质的量。如果样品中存在干扰物质,则浓度的增加值将小于或大于理论值。标准曲线法适用于标准曲线的基体和样品的基体大致相同的情况,优点是速度快,缺点是当样品基体复杂时不正确。标准加入法可以有效克服上面所说的缺点,因为他是把样品和标准混在一起同时测定的(“标准加入法”的叫法就是从这里来的),但他也有缺点就是速度很慢。标准曲线法可在样品很多的时候使用,先做出曲线,然后从曲线上找点,那样方便。而标准加入法,适合数量 少的时候用。在制备标准曲线时,标准液浓度选择一般应能包括待测样品的可能变异最低与最高值,一般可选择5种浓度。浓度差距最好是成倍增加或等级增加,并应与被测液同样条件下显色测定。标准液的测定:在比色时,读取光密度至少读2-3次,求其平均值,以减少仪器不稳定而产生的误差。【来源:实验与分析】
做新药研发的时候,(不算新药,是药物中间体),有些中间体成品需要检测含量。在都有紫外吸收且响应较好的情况下选择外标法测定其含量。有几点疑问,1、何时选择单点含量检测,如果选择单点含量检测,那么这个化合物需要满足的要求是什么,比如选择怎样的响应值合理(即配制浓度选择范围)?2、如果用单点的话,一般对照品和样品如何进样?(我们以前一般是3针对照品取平均值,然后两份样品,各走一针,计算含量,RSD%<2%;但是 逛了一下论坛发现很多都是 走5针或者6针对照,样品平行样各走两针),3、何时选择标准曲线法来计算含量呢?在这个问题下面还有一个前提,是其响应值与浓度比呈线性关系。4、如果响应值与浓度比不呈线性关系呢?那又如何选择检测方法?(试过几个样品,有的样品在一定范围内呈线性,超出范围就不呈线性关系,呈拐点,例如1,2,3个点呈线性,4,5,6呈线性,但是点1和点4却处于平行线上……)如题,欢迎讨论
有人测过硫酸镍中钴的含量吗? 标准加入法和工作曲线法哪种效果好?谢谢啦
石英玻璃中羟基含量检验方法[url]http://www.king-ber.com/Product_Show.asp?ID=755[/url] (红外分光光度计检测)GB/T 12442-901主题内容与适用范围 本标准规定了检验石英玻璃中羟基含量时试样的制备、试验用仪器、试验步骤 及结果处理。 本标准适用于透明石英玻璃中羟基含量的测定。 2引用标准 GB9657 半导体用透明石英玻璃管 GB9658 光源及真空仪表用透明石英玻璃管 JC426-91 无臭的氧石英玻璃管 3术语 3.1透过率或透射比:透过物体的光强度与入射光强度的比值,符号为T。 3.2光谱透过曲线:透过率或透射比随波长的分布曲线。 3.3光密度:衡量玻璃阻止光线透过的能力,符号为D,数值等于透过率倒数的常用对数。 3.4基线:光谱透过曲线上吸收峰两肩边的连线。 3.5零线:光谱透过曲线上透射比为零的线。 3.6摩尔吸收系数:浓度为1mol/L的单位光程长的吸光度,符号为ε。 4试验原理 根据石英玻璃中羟基含量与波长2.73μm处光吸收的线性关系进行定量测定。 5试验仪器 5.1可测波长范围为2.00 ̄3.30μm,测量精度为±1%T的红外分光光度计(TJ270-30A)。 5.2稳定精度为±0.5%的电了交流稳压器。 5.3精度为±0.01mm的千分尺,量度为0.02mm的游标卡尺。 5.4宽度为0.6 ̄1.0mm的长方形固定光栏一组(2个)。 6试样制备 6.1从外观质量符合GB9657、GB9658、JC126或相应标准规定的石英玻璃产品中选取待测样品。 6.2待测样品为块状时,将待测石英玻璃切割、研磨、抛光成镜面,制成25mm×12mm两面平行 厚度差小于或等于0.05mm)的透明试样两个,厚度为0.1 ̄10.0mm,其中人造石英玻璃厚度为0.1 ̄0.7mm,气炼玻璃厚度为0.8 ̄3.0mm,电熔石英玻璃厚度为1.6 ̄10.0mm。使试样在2.73 μm处的透过率在10% ̄80%(吸光度在1.0 ̄0.1)的范围内。 待测样品为管状时,将待测石英玻璃管切取长35 ̄40mm的管段二段,再分别沿管长方向切 取弦长为8 ̄15mm的弧形试样各一个。 6.3用千分尺或卡尺测量试样的厚度。 6.4将试样在器皿中用无水乙醇洗涤干净、晾干、待测。 7试验步骤 7.1开启并预热试验用仪器。当检测管状试样时,在样品光路和参考光路中分别装入长方形 固定光栏。 7.2用遮蔽物遮盖样品光路,盖上样品室的盖,检查并调整仪器零点。 7.3拿去遮蔽物,记录并调整仪器在2.00 ̄3.30μm范围内的100%基准线。 7.4将块状待测试样置于样品光路中,当检测管状样品时,将其试样固定在样品光路的固定 光栏架上,使其中心对准光栏。 7.5按动扫描进行扫描,记录试样在2.00 ̄3.30μm范围的光谱透过曲线。 7.6将试样上下位置对换,再记录一次光谱透过曲线。 8试验结果 8.1在记录的光谱曲线上,划出基线,分别测量出2.73μm处基线到零线和吸收峰到零线的距离。 8.2石英玻璃的羟基含量用下式计算: 1I0 C=96.5──lg──dI 式中:C──试样的羟基含量,ppm; d──试样厚度,cm; I0──2.73μm处基线到零线的距离,mm; I──2.73μm处吸收峰到零线的距离,mm。 本公式由中国建筑材料科学研究院石英玻璃所实测国产石英玻璃中羟基在2.73μm处的克分了 吸收系数ε=80.1L• moL[-1]• cm[-1],根据朗伯-比耳定律导出的。 8.3每个试样用两次计算结果的算术平均值作为该试样的羟基含量值。取两个试样的算术平均值 作为待测石英玻璃的羟基含量值。 8.4参考附录A(参考件)记录和报告试验结果。 附录A 试验记录和试验报告 (参考件) 试验中使用的试验条件应记录在光谱透过曲线的记录纸上,按下述格式和内容填写试验 记录和报告。 石英玻璃羟基含量试验记录 送样单位_____送样日期_____试样名称_____试样数量_____ 试验日期_____试验人员_____ 检验结果 试样编号试样名称试样厚度I0I羟基含量,ppm cmmm检测值平均值石英玻璃羟基含量试验报告 送样单位_____试样名称_____ 试验人员_____报告日期_____ 检验结果 试样编号称试样名称羟基含量,ppm附加说明: 本标准由国家建筑材料工业局提出。 本标准中国建筑材料科学研究院技术归口。 本标准由中国建筑材料科学研究院石英玻璃研究所负责起草并解释。 本标准起草人王明龙。
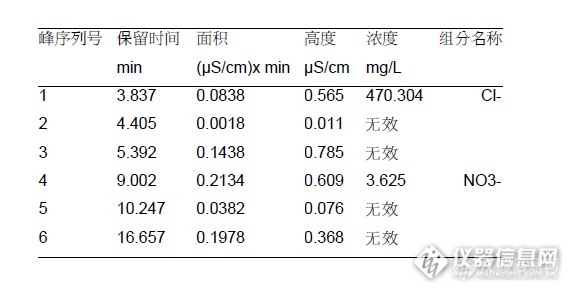
离子色谱法检测矿泉水中硝酸根的含量氮元素是构成生命体的一种重要元素,也是有机物中含量较高的一种元素,在人体内生物代谢有两种途径,其中一种是有机途径,最终产物为尿素,另外一种途径就是无机途径,其代谢的最终产物会是硝酸根。硝酸根本身没有什么危害,但是一旦进入人体,发生氧化还原反应,其危害就暴露出来了。本文中建立了一种使用离子色谱法检测矿物质水中硝酸根离子的方法。该方法能够快速准确的检测出矿物质水中硝酸根离子的含量。仪器设备本文中所使用的离子色谱仪为瑞士万通882离子色谱仪,所配的检测器为电导检测器。使用的色谱柱为Metrosep A Supp 4 - 250/4.0阴离子交换色谱柱,色谱柱的流速为1.0mL/min。淋洗液为Na2CO3和NaHCO3的混合液浓度为1.8mmol/L和1.7mmol/L。色谱的采集时间为20分钟。标准溶液配制称取相当于0.1克硝酸盐含量的硝酸铵,稀释到100毫升容量瓶中,随后移取10毫升稀释液,再次转移到100毫升容量瓶中。此时,分别吸取0.25、0.5、1.0、2.0、2.5毫升二次稀释液,定容到25毫升的比色管中。 http://ng1.17img.cn/bbsfiles/images/2013/12/201312011501_480170_2428063_3.png样品的检测谱图如下,本图中只对硝酸根离子的标准品进行了定量,所以本实验只检测硝酸根离子,检测的结果谱图如下图:http://ng1.17img.cn/bbsfiles/images/2013/12/201312011501_480171_2428063_3.png样品预处理及检测:由于矿泉水样品比较纯净,直接通过0.45um的尼龙微孔滤膜进行过滤处理即可。所使用微孔滤膜由安谱免费赞助,在此表示感谢!根据Magic软件的运算结果,该水样品中硝酸根离子含量为: http://ng1.17img.cn/bbsfiles/images/2013/12/201312011501_480172_2428063_3.png数据显示,百岁山矿泉水中硝酸根离子含量为3.625mg/L,符合国家标准中规定的含量限制,所以,该水样结果为满意结果。结论本文建立了一种离子色谱法检测矿物质水中硝酸根离子含量的方法,所选用的百岁山矿泉水中硝酸根离子含量为3.265ppm,能够达到饮用水中硝酸根的限制,属于合格产品。本实验中,标准曲线的相关系数做到了4个9和一个8是相当的不错啊!表示非常的满意!总体感觉万通的离子色谱做的真不错,赞一个!
色谱买的氧化物标样,还用分析校正其含量吗?不是标准物质吗?
各位气相色谱老师,我刚开始用气相色谱分析气体含量。现在需要测定丙烯/丙烷的标准曲线。但是现在没有标准气体,请教各位老师,一般需要个浓度就能绘制出混合气体的标准曲线?标准气体只需要买一种浓度的再自己稀释还是需要买各个不同浓度的?由于现在实验室经费有限,希望各位老师帮忙建议一个比较合适的方法。谢谢各位老师了!
高效液相色谱测某样品含量标准品用使用内标进行标定吗高效液相色谱测某样品含量,样品处理后要用内标标记,那做标准曲线用的标准品,在制备时除了用流动相溶解,这同时还用使用内标进行标定吗~~
[color=#444444][url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]在线检测氢含量[/color][color=#444444]做氢的标准曲线,这标曲做一次能一直用吗?(在做标曲时,氢气所走的线路必须与反应产生的氢气所走线路相同?)[/color][color=#444444]实际上发现,对于相同含量的氢,每天色谱的相应信号有变化[/color][color=#444444]谢谢[/color]
实验室新购一台波长X荧光光谱仪,需要对岩石,水泥等样进行大量检测,为节约时间,需要用压片法进行常规检测。故需建立压片测试标准曲线。目前遇到的问题如下:1.为消除基体效应,采取以样品本身为标准物质建立标准曲线(以熔融样品前处理检测结果为准)但是会使曲线跨度过小,无法满足检量线两端样品测试2.购买纯物质进行模拟配置相应基体作为标准物质来减弱基体效应的影响,最佳配比量的选择及标准品金属氧化物晶体结构是否会有影响?请各位达人讨论,哪种方案为最佳,或者有更好的方案解决,谢谢!
在做能力验证样品,使用内标法定量。初测发现,目标物含量超过标准曲线线性100倍,你是如何稀释进样的?加大内标浓度后同比例稀释还是先稀释再加内标物?
油品含氧化物含量测定法_气相色谱法_试验报告1前言将被测油品用微量注射器注入气相色谱仪,采用中心切割技术 ,使样品从第一根色谱柱DB-1(非极性柱)预分离后的部分馏分,被切割到第二根色谱柱Lowox(极性柱)以进一步进行分离,两根柱子通过特殊的装置连接起来,允许一个或多个感兴趣的组分从第一根柱转移到第二根柱,两根柱间的切换是通过气流控制开关来实现。由于两根柱的极性不同,在第一根柱中共流出的含氧化物组分在第二根柱中得到完全分离。通过氢火焰离子化检测器检测,采用外标法对各含氧化物进行定量。中心切割阀路连接示意图,见图1http://ng1.17img.cn/bbsfiles/images/2014/12/201412201741_528063_2266096_3.png2 实验部分色谱条件:安捷伦气相色谱7890A; 色谱柱1:DB-1非极性柱;30m*0.53mm*2.65um;色谱柱2:Lowox极性柱;10m*0.53mm*10um双检测器:FID 250℃;进样口:200℃色谱柱温度:100℃(5min) 5℃/min 130℃ 10℃/min 225℃(4.5min)2.2 校准曲线绘制称量干净的100mL空容量瓶,加入异辛烷到刻线,称量得出异辛烷的量为70.1781g,依次加入MTBE、ETBE、甲醇、乙醇各约50µL,称量得出各含氧化物的量为:MTBE 0.0374g、ETBE 0.0432g、甲醇 0.0408g、乙醇 0.0391g,计算各含氧化物的浓度为:MTBE 532.9 mg/kg、ETBE 615.5 mg/kg、甲醇 581.3mg/kg、乙醇 557.1mg/kg。对上述溶液进行稀释,得到表1的一系列标准样品。http://ng1.17img.cn/bbsfiles/images/2014/12/201412202019_528085_2266096_3.pnghttp://ng1.17img.cn/bbsfiles/images/2014/12/201412202021_528086_2266096_3.pnghttp://ng1.17img.cn/bbsfiles/images/2014/12/201412202021_528087_2266096_3.pnghttp://ng1.17img.cn/bbsfiles/images/2014/12/201412202021_528088_2266096_3.pnghttp://ng1.17img.cn/bbsfiles/images/2014/12/201412202021_528089_2266096_3.pnghttp://ng1.17img.cn/bbsfiles/images/2014/12/201412202025_528092_2266096_3.pnghttp://ng1.17img.cn/bbsfiles/images/2014/12/201412202025_528093_2266096_3.png
为什么说低含量的样品对标准曲线线性要求更高?
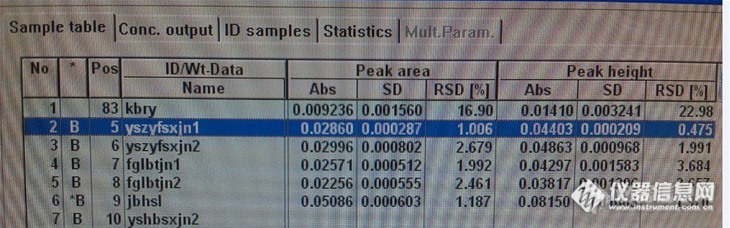
前几天做铬,公司给定的sop上面的标准曲线点事0,10,20,30ng/ml,样品的处理是0.5g样品加10ml硝酸,浸泡过夜,第二天消解,赶酸,稀释至50ml,上机测试,样品中铬的含量一般在0.1-0.2mg/kg之间。 我每次看样品溶液的吸光度都在曲线的最下部分,因为之前看论坛里某些老师说过,被测溶液的吸光度值最好在标准曲线的中部,然后我想想,我这样是不是可以?结果是否准确? 然后我就动手了, 称了两份样品,顺便做了个加标回收,配了2套标准曲线,第一套按照SOP 标准配的,第二套标准曲线点位0, 1.0, 2.0, 4.0, 10.0ng/ml 然后开始测试。仪器条件如下,http://ng1.17img.cn/bbsfiles/images/2013/06/201306161827_445595_2575221_3.jpg 第一套曲线测定如下:http://ng1.17img.cn/bbsfiles/images/2013/06/201306161828_445596_2575221_3.jpg第一套标准曲线测定结果如下:吸光度:http://ng1.17img.cn/bbsfiles/images/2013/06/201306161843_445602_2575221_3.jpg计算得结果如下:http://ng1.17img.cn/bbsfiles/images/2013/06/201306161831_445597_2575221_3.jpg第二套标准曲线测定如下:http://ng1.17img.cn/bbsfiles/images/2013/06/201306161832_445598_2575221_3.jpg按照第二套标准曲线重新计算得出结果如下:http://ng1.17img.cn/bbsfiles/images/2013/06/201306161834_445599_2575221_3.jpg注:样品表格中编号2,3为被测样品,1为空白溶液,6为加标回收空白溶液为样品空白,即加10ml硝酸,与样品同时同样处理加标回收:样品取样量0.5101g,加入100ul标准溶液(浓度为1ug/ml),加硝酸10ml 过夜,消解,赶酸,稀释至50ml。(1)大家认为要是您测定铬,您选哪套标准曲线,原因是什么?(2)大家算算加标回收率多少?
液相测含量为什么不做标准曲线
[size=5]超临界流体色谱快速测定烟酰胺的含量[/size] 来源: 作者:郭亚东,马银海,张艳,彭永芳摘要:采用超临界流体色谱快速测定制剂中烟酰胺的含量.在CO2流动相中添加10%的甲醇,于填充柱上分离,检测波长为216nm,在测定范围内,浓度与其峰面积呈良好的线性关系(r=0.9998),峰面积的相对平均偏差(RSD)为1.39%,平均回收率97.3%~101.3%,4min即可完成分析.方法简便,样品前处理简单,可用于制剂中烟酰胺的快速分析。关键词:烟酰胺;超临界流体色谱;含量测定水溶性维生素对人们的生长发育和健康有着重要作用,人们除了从水果和蔬菜中摄取外,还从添加了维生素的食品和复合维生素制剂中补充人体的需要,有必要建立快速,稳定的分析方法用于其含量测定。对烟酰胺的含量测定方法主要是高效液相色谱法,该方法取得了较好的结果,但其分析时间长,样品前处理麻烦。此外,毛细管电泳,胶束电动毛细管电泳,气/质联用等方法也用于它的含量测定.本文探索了用超临界流体色谱测定维生素含量的方法,结果令人满意。1 仪器与试药超临界流体色谱仪:Gihon Model SF3系统(英国),对照品烟酰胺购自Lancaster化学公司(英国),甲醇为高效液相色谱纯,CO2为超临界流体色谱纯.2 实验方法及结果分析2.1 色谱条件色谱柱cyano(5μm,4.6×250mm),柱温50℃,紫外检测器配有高压检测池,其检测波长为216nm,进样装置带有l0μL进样阀的自动进样器,流动相压力20MPa,流动相流速为2.0mL/min。2.2 流动相对分离的影响只用CO2作流动相时,其保留时间太长,且色谱峰拖尾严重;当在流动相中加人10%的甲醇后,其峰形大为改善,保留时间缩短;当流动相流速改变时,保留时间会有小的改变,但对峰形几乎没有影响。2.3 线性关系考查将烟酰胺对照品取适量,精称后用甲醇稀释成5~50μg/mL的标准溶液,取6个不同浓度的对照品溶液按上述实验条件各进样三次,记录其色谱图,以对照品浓度(μg/mL)为横坐标,峰面积值为纵坐标,绘制标准曲线,得回归方程Y=一351.6+147.7X,R=0.9998,可见在所用浓度范围内具有良好的线性关系。2.4 精密度及稳定性试验测定该维生素日内和日间的峰面积,以考查分析方法的精密度和样品的稳定性,在上述实验条件下,连续进样8次,烟酰胺峰面积的相对标准偏差(RSD)为1.11%,放置1d后的RSD为1.39%,表现出良好的精密度和稳定性。2.5 回收率试验精密称取已知含量的同一样品三份,加人不同量的烟酰胺对照品,按样品测定项下的条件进样分析,计算其回收率,烟酰胺的回收率平均值±SD为98.3±1.28%。2.6 样品测定取昆明振华制药厂生产的复合维生素B(批号990701)5片,碾碎后用5mL甲醇溶解并超声提取20min,用0.45μm滤膜过滤,适当稀释后在上述色谱条件下进样分析,以峰面积按标准曲线法计算含量。3 讨论3.1 流动相选择当用CO2加10%甲醇作流动相时,烟酰胺的保留时间为3.7min。可以满足快速分析的要求,且色谱峰形得到改善。3.2 提取溶剂的选择比较了水、甲醇和乙醇作溶剂提取样品,结果以甲醇较好。样品用甲醇溶解并超声提取后,直接进样分析,不需要复杂的前处理.3.3 结果通过测定回收率,精密度并考查其线性关系,表明该方法可以于快速测定烟酰胺含量,能给出满意的结果.[参考文献][1] Hurtado S A,Nogues M T V,Pulido M I et a1.Determination of water—soluble vitamins in infant milk by HPLC[J].chromato A,1997,778:247.[2] Wills R B H,Shaw C G,Day W R.Analysis of water soluble vitamins by PHLC[J].Chromatogr Sci.,1977,15:62.
请教如何理解多点内标法这一制作标准曲线的问题。我的理解是这样的,不知可否正确,烦请专家指正。由于内标法要求待测物含量要与内标物含量相当,如果待测物含量有一定的范围的话,为了准确对其定量,则要求内标含量也需与之相当,从而就需配制含有不同内标量不同待测样品量的标准溶液系列(比如5个)。用这些样品在GC上分别用单点内标法测定而得到5个独立的文件,作为多点内标法校正曲线上5个点的来源。
最近在使用[url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]测定乙醇含量,在采用外标法做标准曲线时遇到问题:相同浓度的乙醇标准样进样后得到峰面积却相差很大,我重复5次进0.2μl标准样时得到的峰面积为:8840,3904,8434,7563,14712,想请教下各位是什么原因!







 我要推广仪器
我要推广仪器
 下载APP
下载APP