高分子表征技术专题——拉曼光谱技术在高分子表征研究中的应用
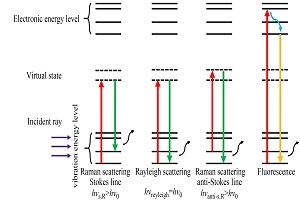
2021年,《高分子学报》邀请了国内擅长各种现代表征方法的一流高分子学者领衔撰写从基本原理出发的高分子现代表征方法综述并上线了虚拟专辑。仪器信息网在获《高分子学报》副主编胡文兵老师授权后,也将上线同名专题并转载专题文章,帮助广大研究生和年轻学者了解、学习并提升高分子表征技术。在此,向胡文兵老师和组织及参与撰写的各位专家学者表示感谢。更多专题内容详见:高分子表征技术专题高分子表征技术专题前言孔子曰:“工欲善其事,必先利其器”。 我们要做好高分子的科学研究工作,掌握基本的表征方法必不可少。每一位学者在自己的学术成长历程中,都或多或少地有幸获得过学术界前辈在实验表征方法方面的宝贵指导!随着科学技术的高速发展,传统的高分子实验表征方法及其应用也取得了长足的进步。目前,中国的高分子学术论文数已经位居世界领先地位,但国内关于高分子现代表征方法方面的系统知识介绍较为缺乏。为此,《高分子学报》主编张希教授委托副主编王笃金研究员和胡文兵教授,组织系列从基本原理出发的高分子现代表征方法综述,邀请国内擅长各种现代表征方法的一流高分子学者领衔撰写。每篇综述涵盖基本原理、实验技巧和典型应用三个方面,旨在给广大研究生和年轻学者提供做好高分子表征工作所必须掌握的基础知识训练。我们的邀请获得了本领域专家学者的热情反馈和大力支持,借此机会特表感谢!从2021年第3期开始,以上文章将陆续在《高分子学报》发表,并在网站上发布虚拟专辑,以方便大家浏览阅读. 期待这一系列的现代表征方法综述能成为高分子科学知识大厦的奠基石,支撑年轻高分子学者的茁壮成长!也期待未来有更多的学术界同行一起加入到这一工作中来.高分子表征技术的发展推动了我国高分子学科的持续进步,为提升我国高分子研究的国际地位作出了贡献. 借此虚拟专辑出版之际,让我们表达对高分子物理和表征学界的老一辈科学家的崇高敬意!拉曼光谱技术在高分子表征研究中的应用Application of Raman Spectroscopy in the Characterization of Polymers作者:袁媛,王梦梵,曲云菲,张泽军,张建明作者机构:青岛科技大学高分子科学与工程学院 橡塑材料与工程教育部重点实验室,青岛,266042 北京化工大学 碳纤维及复合材料教育部重点实验室,北京,100029 北京航空航天大学化学学院,北京,100191作者简介:张建明,男,1973年生. 山东省泰山学者特聘教授,博士生导师. 2003年毕业于中科院化学所并取得博士学位,师从著名的光谱学家沈德言先生. 自2009年1月起在青岛科技大学工作. 研究方向为高分子凝聚态结构及其相变行为、生物质纳米材料制备及功能复合材料构筑,已发表SCI学术论文130余篇,所发论文被引6000余次,H-指数为38,获批中国发明专利20余件. 先后获日本JSPS博后奖、德国洪堡资深学者、山东省泰山学者、山东省杰出青年、山东省自然科学二等奖及中国石化联合会青年科技突出贡献奖等荣誉或奖励.摘要拉曼光谱作为一种强大的工具,被广泛应用于聚合物结构的表征. 随着共振拉曼光谱、扫描角度拉曼光谱、高分辨率拉曼成像、极化拉曼光谱、表面增强拉曼散射等拉曼技术的迅速发展,拉曼光谱的应用范围不断扩大. 本文首先介绍了拉曼光谱设备的基本原理和组成,总结了拉曼技术的实验技巧和数据处理中需要注意的问题,讨论了红外光谱和拉曼光谱的区别,在此基础上,综述了近十年来拉曼技术在聚合物结构表征领域的最新应用和研究进展. 其应用包括以下六个方面:高分子链的构象、聚合物的聚集状态、聚合物结晶度的计算、高分子链的取向、外场作用下的结构转化、高分子共混物化学或物理成分的识别. 最后,对拉曼光谱在聚合物研究中的发展进行了展望. 希望本文能够对试图从拉曼光谱中获取聚合物结构信息的学者有所帮助.AbstractAs a powerful tool, Raman spectroscopy is widely used in the characterization of polymer structures. Along with the rapid development of Raman technology such as resonance Raman spectroscopy, scanning angle Raman spectroscopy, high-resolution Raman imaging, polarized Raman spectroscopy, and surface-enhanced Raman scattering, the application range of Raman spectroscopy has been continuously extended. In this paper, we first introduced the basic principle and the composition of the Raman equipment, and then we summarized the experimental skills of Raman technology and the issues that need attention in data processing. The difference between the infrared spcectroscopy and the Raman spectroscopy was discussed. Afterwards, we reviewed the latest applications and research progress in the fields of polymer structure characterization by using Raman technology in recent decade. The applications include the following six aspects: the macromolecular chain conformation, the aggregation state of polymers, the calculation of the polymer crystallinity, the macromolecular chain orientation, the structural transformation under the external fields, and the identification of the chemical or physical composition in polymer blends. Last, the development of Raman spectroscopy in polymer research was prospected. It is hoped that this review could be helpful for the one who tried to obtain the information about the polymer structure from Raman spectroscopy.关键词拉曼光谱 结构表征 原理 应用KeywordsRaman spectroscopy Structure characterization Principle Application 拉曼散射现象是由印度科学家Raman于1928首先发现并报道的,但拉曼散射信号只相当于瑞利散射百万分之一,在拉曼散射现象被发现之初由于没有足够功率的光源而并未被广泛的应用. 近半世纪以来随着激光光源以及显微技术在拉曼光谱仪中的应用,拉曼光谱迸发出了旺盛的生命力.拉曼光谱与红外光谱同属分子振动光谱,但其原理与红外光谱截然不同. 如今拉曼光谱在高分子领域中已经有广泛的应用,包括分子链构象、取向、结晶度等方面的研究等. 本文在结合拉曼基本原理及实验技巧的基础上,总结了近年来拉曼光谱在高分子表征中的最新研究进展.1基础原理1.1光的散射当光线遇到分子时,绝大部分的光子(多于99.999%)都会发生弹性散射(即瑞利散射),瑞利散射具有与入射光相同的波长. 然而,少部分的光子(少于0.001%)会发生能量(频率)偏离的非弹性散射(即拉曼散射). 光散射过程可以用量子力学进行描述,如图1所示,当一束光照射到某体系时,体系中粒子吸收光的能量而被激发,从而发生能级跃迁过程,同时辐射出散射波. 不同的跃迁方式决定了不同的散射类型,例如(拉曼)斯托克斯散射、瑞利散射、(拉曼)反斯托克斯散射(高分子样品测试中常用的拉曼散射范围)[1~7]. 在拉曼测试过程中,经常也会出现荧光信号,与拉曼散射不同,荧光过程中粒子被激发至能量更高的电子能级而非拉曼散射中的虚态. 因此短波长比长波长激光更易产生荧光效应.Fig. 1Quantum mechanics description of Rayleigh, Raman scattering and florescence.1.2拉曼散射与拉曼光谱1.2.1拉曼散射的基本原理假设一束频率为v0的光照射在一个分子上,分子中电子会被入射光的电场激发做受迫局域运动而出现极化现象,产生电偶极矩,假设入射光电场可以表示为:式中E0为光电场的振幅,则由于分子运动所产生的偶极矩可以表示为:式中α为极化率,极化率的变化是分子的核外电子云受外部电场诱导而产生的(通过平衡位置两边的)形变而导致的.如果分子的极化电场所释放出的光与入射光频率相同,则把这种散射过程称为瑞利散射. 而如果α被分子的振动所调制(modulated),则α可以展开为关于振动简正坐标q的级数:q由以下公式得出:则有:以上公式表明在当前情况下频率为(v0±vk)的(拉曼)散射会与频率为v0的瑞利散射同时出现. 某一分子振动为拉曼散射活性的前提条件为(∂α∂q)0的值不为0,也就是说分子的极化率随分子振动而改变[8,9].如图2所示,假设频率为v0电场(入射光)可以诱导分子的偶极矩P产生同频率(v0)的振动. 如果此时分子极化率具有随时间变化的极低频的振动vm,那么经过以上2种不同频率的振动调制后的散射光将包含3种不同频率的光,分别为v0(瑞利散射)、v0+vm(反斯托克斯散射)、v0-vm(斯托克斯散射). 反之如果分子的振动不能使极化率产生低频振动,则不会有调制的出现,进而不会出现拉曼散射效应[8,10].Fig. 2Schematic representing of Rayleigh and Raman scattering: (a) the incident radiation makes the induced dipole moment of the molecule oscillate at the photon frequency (v0) (b) the molecular vibration can induce the polarizability,α,to have a frequency ofvm the result as shown in (c) is an amplitude modulated dipole moment oscillation,and three components with steady amplitudes which can emit electromagnetic radiation can be achieved as:v0 (Rayleigh component), v0+vm (Raman anti-Stokes component), and v0+vm (Raman Stokes component), as shown in (d).由于诱导分子偶极矩P与电场E均为矢量,且一般情况下两者方向不同,因而连接这2个物理量的极化率α可以用一个二阶张量来表达,则P=αE可以表示为其中,x,y,z为分子在笛卡尔坐标系中的坐标. 极化率为对称的二阶张量矩阵,包含了6个独立的元素,αxx、αyy、αzz、αxy、αyz、αxz. 上式的意义为,例如沿x方向电场Ex诱导了沿y方向的偶极矩Py,则可表示为Py=αxyEx. 此式在通过偏振拉曼研究分子对称性时具有重要意义[9].1.2.2拉曼活性的判据如上所述,非弹性散射源于在平衡位置附近分子的极化率关于简正坐标q的导数不为0,这一关系为小分子的拉曼散射提供了“选择定律”的基础. 以对称双原子分子的对称伸缩振动(symmetric stretching vibration)为例,如图3(a)所示,当两原子的位置无限接近时,体系电子密度分布类似于单一原子的电子密度;而当两原子的位置无限远离时,体系电子密度分布近似于2个独立的单原子的电子密度. 因此对于双原子分子的对称振动,其极化率沿简正坐标方向成单调增长模式,因此其在平衡位置导数不为0,为拉曼活性振动. 而对于分子偶极矩,对称伸缩振动过程中其正负电荷中心并没有产生位移,所以偶极矩没有发生变化,因此为红外非活性振动. 例如氧气与氮气分子的对称伸缩振动只能使用拉曼光谱进行研究,因为在红外谱图中不会出现吸收峰.Fig. 3The derivatives of polarizability (red) and dipole moment (blue) are schematically depicted for the normal modes of a two (a) and a three (b) atomic molecule. Based on these intuitive considerations,conclusions on the IR and Raman activity of the modes can be drawn.线性三原子分子比双原子分子稍显复杂,例如二氧化碳分子. 对于其对称伸缩振动,如图3(a)所示,极化率的变化类似于双原子分子的对称伸缩振动,为拉曼光谱活性,红外光谱非活性. 对于非对称伸缩振动(antisymmetric stretching vibra-tion)以及变角振动(bending vibration) (图3(b)),极化率在平衡位置两边的变化虽不为0,但是其变化是关于平衡位置对称的. 因此极化率在平衡位置周围变化可以认为是简谐的,也就是说(∂α∂q)q0=0,因此非对称伸缩振动与变角振动均为拉曼非活性;而偶极矩在平衡位置两侧的方向是反转的,因此(∂μ∂q)q0≠0,表现为红外活性[11].2实验技巧为了得到更丰富的样品信息,我们希望拉曼光谱在准确的基础上具有尽可能高的信噪比(signal-noise ratio,SNR). 关于拉曼散射的强度IR一般有如下关系式:其中,v和I0为入射激光的频率及强度;N为参与散射过程的分子数量;(∂α∂q)2是与分子结构有关的参数.上式表明,使用短波长激光并增加激光能量密度的同时增加样品量可以增强拉曼散射信号(注:拉曼光谱位移不随入射波长的变化而改变). 但在实际的测试过程中,不同类型的样品需要根据其自身的特点选择与其匹配的波长的激光以及激光能量,不能为了增强拉曼信号就去用短波长激光去测试所有样品,很多高分子样品在短波长激光下可能没有拉曼信号或者拉曼散射被很强的荧光信号所淹没.2.1样品制备2.1.1固态样品相对于无机样品,有机高分子样品的拉曼信号相对较弱(一部分原因是由于高分子样品中存在大量的无序结构). 对于高分子粉末或膜样品,一般需要保证沿光的入射方向有一定的厚度并同时使其表面尽量平整,以便于显微镜的聚焦. 对于透明样品,可将其放置于铝箔上进行测试(因为金属一般都有增强拉曼信号的作用,用铁片作为基底同样有着很好的效果). 或者,由于拉曼接收的是散射光,太薄的透明样品极易被激光穿透从而打到基底上,因此为了得到更好的拉曼信号,制样时要尽可能增大薄膜厚度. 另外由于激光一般都是偏振的,因此对于取向样品,例如纤维,需首先确定入射光的偏振方向,之后再确定样品的(某一)取向轴与入射光偏振方向平行(或垂直),再开始测试,这样才能得到正确的结构信息.2.1.2液态样品由于拉曼可以聚焦到几十微米下检测一定深度的样品信号,无需担心盖玻片和毛细管对拉曼信号的影响,因此高分子液态样品的拉曼测试相对于红外测试比较便捷,可以直接进行测试. 一般可以使用凹面载玻片或者金属制液体样品槽承载液体样品. 测试时可先将激光聚焦于液体表面,然后将样品平台沿激光方向上抬,使激光聚焦于液体样品内部,这样可以得到较好的光谱. 如果液体易挥发,可以使用盖玻片将样品封闭于容器内或将液体封入毛细管内.2.2设备调试2.2.1拉曼装置的构成随着拉曼仪器的发展,如今在一般情况下,背散射模式,也就是入射激光与散射激光平行,已经足够应对大部分高分子样品的测试需求. 对于一些特殊情况,例如取向或单晶样品的偏振拉曼测试,需要使用到90°入射的模式,也就是入射光路方向与散射光路方向为90°,原因可以参考上节极化率的二阶张量公式.以雷尼绍(Renishaw,UK) inVia型拉曼光谱仪为例,如图4所示,拉曼装置一般包括入射激光光源、入射光路系统(包括扩束器)、显微镜及样品台系统、滤波器、衍射光栅及CCD检测器. 在实际测试过程中,我们需要选择合适的入射光波长及显微镜物镜.Fig. 4Schematic diagram of the Raman instrument.当今市场上主要的拉曼仪器根据应用的场景可分为手持型、便携型以及桌面型拉曼光谱仪. 手持型拉曼光谱仪集成性很高,小巧轻便,操作非常简单,几乎可以在各种需要的地点、时间对从原材料到成品进行鉴定分析. 便携型拉曼光谱仪集成性相对较高,并具有一定的扩展性,可作为小型移动实验室使用. 桌面型拉曼光谱仪体积较大且不可移动,如图4中示意图即为桌面型拉曼光谱仪,但这类光谱仪具有极强的扩展性,几乎可以变更从入射激光光源、入射光路、样品平台至光栅等所有组成部分,从而可以为不同样品以及不同条件的测试创造可能.2.2.2激光波长的选择激光波长与能量密度成反比,使用短波长激光可以得到较强的拉曼散射信号,例如532 nm要比785 nm激光的拉曼散射强度强. 但对于高分子样品来说使用532 nm激光产生荧光干扰的可能性也会增加. 所以在一些情况下可以选择785 nm的光源. 如前所述,样品产生的拉曼位移不会随激发光源的波长改变而改变,因此只要可以避开荧光效应可以自由选择激光波长. 需要注意,虽然拉曼位移不随激光波长而改变,但使用同一物镜下,不同波长可以到达的空间分辨率不同. 例如,物镜的数值孔径(NA)为0.9,532 nm激光的空间分辨率可达0.72 μm,而在同样条件下使用785 nm激光时,空间分辨率仅为1.1 μm.另一种情况,如果样品内的分子振动与入射激光可以产生共振效应,那么可以以此来选择入射激光波长,则可以得到较强的拉曼散射信号.2.2.3显微镜的选择通常显微镜的物镜上会标注2个参数,分别为放大倍数(5×、10×、20× 等)与数值孔径(numerical aperture,NA,是与镜头光通量有关的参数,一般为0.05~0.95). 一般放大倍数与数值孔径成正相关关系,而数值孔径决定空间分辨率,有如下公式 [12]:其中,R为最大空间分辨率. 在实际测试时需要注意激光能量会随光斑尺寸(空间分辨率)变化,更高的空间分辨率意味着激光密度会更大,此时需要注意样品可能会被激光热解. 对于高分子样品来说,一般要先从低激光功率测试开始尝试,如果此时拉曼散射信号很弱,则少量增加激光功率,但同时要注意观察样品是否被热解,如此反复尝试直到找到最适宜测试的激光强度.2.2.4Ne灯校准一般除用单晶硅对拉曼位移进行校准,另外使用内置的Ne灯也可以达到校准的效果. 一般在测试样品时与Ne灯同时使用,则所得到的拉曼谱图中同时包括样品与Ne灯的峰,由于Ne灯的拉曼峰位置已确定,因此可用于校正样品的峰位置.2.2.5测试参数设置在确定适宜样品的激光波长及显微镜倍数的前提下,为了提高信噪比,可以首先在不损伤样品的前提下尽量提高入射激光的强度,其次适当延长曝光时间(有效的提高散射信号强度),同时也可以增加循环(cycling)测试的次数(有效降低噪音的影响). 但需要注意曝光时间不宜过长,因为过长会导致检测器的饱和,例如当同时需要较强与较弱的拉曼散射峰时,较弱的散射峰由于信噪比较低而难以使用时,可以固定曝光时间并增加循环测试次数来降低最终谱图中噪音的干扰.2.3数据处理2.3.1高分子样品拉曼谱的初判在取得拉曼光谱后,首先需要对谱图的构成进行判断,因为其中可能同时包含样品以及非样品的拉曼信号. 如果可以排除样品不纯净的可能,那么非样品的拉曼信号可能来自于宇宙射线、自然光或照明光等所产生的干扰,另外如果样品透光性好,激光可能透过样品打到基底上,也可能产生部分非样品信号.宇宙射线所产生的特征峰强度高且十分尖锐,并且可能在任意波数出现. 而如果在测试时对照明光抑或显示器背光的屏蔽不彻底,则也会出现一些尖锐的谱峰,这些谱峰的位置与光的类型有关. 但同宇宙射线不同的是,这些峰不是随机出现,而是会在相同的位置重复出现.对于结晶性高分子样品来说,由于内部存在大量的晶格缺陷及非晶组分,通常即使是结晶特征峰也不会是非常尖锐的峰,这种情况类似于红外测试的结果. 一般来说,对于同一振动模式,相较于非晶峰,结晶峰的峰强较强,峰宽较窄. 对于未知的结晶性高分子样品,可以通过分别测试结晶与熔融状态下的样品来确定结晶与非晶的特征峰. 确定特征峰是进一步测试分析的基础.由于我们常规使用的拉曼散射的波束范围恰好与中红外测试波段相似(400~4000 cm-1),并且两者均为分子的基团振动光谱,所以兼具红外与拉曼活性的同一分子基团振动在两谱图中的频率相似,两者可以互为参考. 而在低波数范围(400 cm-1),也就是远红外区间(一般反应分子链主链的振动),由于空气中的水气对测试有极大的干扰,所以远红外测试需要对样品仓抽真空,这也极大地限制了远红外光谱的应用,因此在实际测试中远红外与中红外区不能同时测试. 而拉曼的测试范围可以直接覆盖远红外及中红外波束段,并且测试过程中无需进行硬件切换,这也为高分子的研究提供了极大的便利.2.3.2谱线的平滑与拟合在一些情况下,由于样品或仪器的原因,即使已经选择了最优的测试条件,所得的光谱仍可能存在信号起伏大,信噪低的情况. 此时为了便于数据分析,可以对光谱进行平滑或拟合处理. 但是由于平滑后光谱会发生微小的变化,例如肩峰可能会因此消失,所以在对样品光谱没有十足把握的情况下,进行平滑处理时要十分谨慎. 一般如果噪声水平在中整条光谱中都比较均一,可以对光谱进行平滑处理,在平滑时,尽量选用最少的数据点个数为平滑单位,不能以牺牲数据准确来换取谱线的平滑美观. 在其他情况下,例如存在非拉曼信号,则不能使用平滑处理来消除,而应改变测试条件来避免非拉曼信号的产生.当谱图中有2个或多个峰重叠时,为了便于分析数据,需要进行分峰拟合(通常使用高斯加洛伦兹函数拟合),要注意虽然拟合的目标是尽量还原原始光谱,但不能为了达到这个目标而任意增加分峰的个数而忽略了每个峰的物理意义,这样便失去了分峰的价值.总之,不论何时原始数据都是最重要的,任何数据处理方法都需要在遵从原始数据的基础上进行.3拉曼光谱应用举例2010年至今,拉曼光谱在高分子多层级结构解析中的应用主要涉及6个方面,分别是:分子链构象研究、分子聚集态研究、结晶度计算、分子链取向研究、外场作用下的结构转变研究、化学/物理组成研究. 应用到的拉曼光谱种类主要为:共振拉曼光谱(resonance Raman spectro-scopy)、扫描角度拉曼光谱(scanning angle Raman spectroscopy)、高分辨拉曼成像(high-resolution Raman imaging)、偏振拉曼光谱(polarized Raman spectroscopy)及表面增强拉曼光谱(surface-enhanced Raman scattering, SERS).3.1分子链构象研究Gao等[13]利用共振拉曼光谱识别了聚(2,5-双(3-十四烷基噻吩-2-基)噻吩[3,2-b]噻吩)(PBTTT)与电子受体[6,6]-苯基C61丁酸甲酯(PCBM)共混的体异质结太阳能电池中PBTTT的有序和无序构象. 作者提出PBTTT噻吩环C=C对称伸缩振动(νs(C=C))包括主链有序构象和无序构象2个组分的贡献:如图5所示,有序构象的特征峰位置在1489 cm-1,半峰宽约为15 cm-1;无序构象的特征峰位置在1500 cm-1,半峰宽约为25 cm-1. PBTTT不同构象的相对含量随PCBM含量、退火温度与拉曼激发能的改变而变化. 共振拉曼图像进一步证实有序的PBTTT链集中在富含PCBM的双分子晶体中. Martin 等[14]同样借助共振拉曼光谱结合光电流成像技术,考察了高分子-富勒烯共混物中依赖于构象变化的电荷沿主链的传输特性. 实验及理论计算的结果均证实当共轭高分子的主链呈现平面构象时,电荷传输率最高. 体系形貌表征的结果表明当高分子与富勒烯达到良好共混状态时,高分子主链构象更易于平面化.Fig. 5Simulated Raman spectra (a) of the BTTT-C2 monomer and structures (b) (Reprinted with permission from Ref.[13] Copyright (2014) American Chemical Society).原位共振拉曼表征被成功地应用于研究ps尺度上聚(3-己基噻吩)(P3HT)分子链在氯苯中的构象松弛过程[15]. 如图6(a)所示,基于激发态拉曼特征的时间依赖性及与其他高分子的拉曼光谱进行对比,作者归属了构象松弛过程中不同结构的拉曼特征峰. 通过绘制拉曼特征峰的强度变化对时间的关系曲线(见图6(b)),揭示了松弛过程中主链共轭长度的变化,据此提出了P3HT分子链在氯苯中的构象松弛动力学机理.Fig. 6(a) Valence-bond structures of the quinoidal excited state of P3HT and the time-resolved resonant-Raman spectra of P3HT in chlorobenzene photoexcited at 510 nm. (b,c) Time dependence of Raman band intensities in figure (a). Integrated intensities (b),black lines correspond to biexponential fits with constrained lifetimes of (9±1) and (220±20) ps. Relative change in feature intensities attributed to torsion-induced exciton conformational relaxation (c). (Reprinted with permission from Ref.[15] Copyright (2012) American Chemical Society).3.2分子聚集态研究Gao等[16]在对P3HT/PCBM共混薄膜分子聚集态的研究中区分了不同聚集态对P3HT主链C=C伸缩振动νs(C=C)的贡献. 对样品光谱的拟合结果(如图7(a)所示)表明,共混膜的(νs(C=C))峰来自于聚集分子链与非聚集分子链的双重贡献,前者的特征拉曼频率约为1450 cm-1,后者约为1470 cm-1. 聚集态与非聚集态峰强度的相对比值R(R = IC=Cagg/IC=Cun)在样品退火后增加(如图7(b)所示),R值与不同聚集态的相对密度相关. 如图7(c)所示,作者进一步应用共振拉曼成像来考察R值变化对共混形貌的依赖关系,通过R值对比,对退火的共混薄膜中4种聚集程度不同的P3HT分子链进行了识别与成像分析. 在此工作基础上,作者通过分析拉曼特征峰的强度变化,考察了P3HT聚集态对共混物体系中局部光电流产生效率[17]、激发态结构变化及初期振动动力学[18]的影响. 共振拉曼结合成像技术分析也被成功地应用于其他共轭聚合物结构与性能的对应关系研究中[19].Fig. 7(a, b) Raman spectra of as-cast (a, red) and annealed (b, blue) blend films excited with 488 nm light show theνs(C=C) band of P3HT represented by the shaded regions of the complete spectra shown as insets. The band is fitted with two Lorentzian functions (dashed traces), showing the relative contributions of both aggregated (IC=Cagg) and unaggregated (IC=Cun) components. (c1 and c2)IC=CaggandIC=Cuncenter frequency dispersion images for P3HT/PCBM as-cast films,and (c3) histograms of frequency components. (c4 and c5)IC=Cagg and IC=Cuncenter frequency images for P3HT/PCBM annealed films,and (c6) histograms of frequency components (Reprinted with permission from Ref.[16] Copyright (2012) American Chemical Society).拉曼光谱结合高空间分辨成像技术可用于高分子多晶型结构,例如针对聚己二酸丁烯酯(PBA)的环带球晶研究[20]. 在此工作中作者首先识别了2种晶型(α晶与β晶)及非晶结构的拉曼特征峰,选择能够反映不同聚集态相对含量的特征峰(C-peak),在此基础上通过拉曼成像考察了球晶内部多晶型晶体的分布及分子链取向. 通过对比球晶的偏光照片(图8(a))与拉曼成像照片(图8(b))可知,2种晶型的晶体在球晶中心、环带区域及外层非环带区域呈现非均匀分布,二者能够在相同的温度区间(31~33 ℃)成核和生长,然而环带区域α晶的相对含量会随结晶温度而提高. 2种晶型的拉曼成像数据结合Hermans取向函数分析(见图8(c))结果证实,环带区域的分子链沿球晶半径方向和基底平面取向,且沿环带球晶径向方向的取向呈周期性变化.Fig. 8(a) Optical micrographs of PBA31-33. (b) Raman imaging of C-peak position for the same area in (a). (c) Hermans orientation function image calculated by using the C-peak area of PBA32 measured with polarization parallel (0°) and perpendicular (90°) to the horizontal direction (Reprinted with permission from Ref.[20] Copyright (2017) American Chemical Society).拉曼成像技术作为一种强有力的表征手段,可以精确表征(分辨率最高可达0.1 μm)单片层石墨烯或氧化石墨烯在片层不同区域的氧化结构. Zhang等[21]通过拉曼成像技术对具有不同氧化结构的单片层氧化石墨烯进行了表征,通过D/G峰的比值差异分析了单片层在不同区域的氧化程度. 如图9所示,JGO纳米片与GO差异显著,后者呈现出统一的颜色(图9(a)和9(b)). 此外,从图9(c)和9(d)可以看出,在蓝色区域(低氧化区域),JGO的ID/IG比值较低(~0.72),而在红色区域(高氧化区域),ID/IG比值较高(~1.07),与GO的ID/IG比值存在显著差异(整个区域的ID/IG比值为~1.02). Badi等[22]同样借助拉曼成像技术,通过D峰与G峰的光谱解析,考察了石墨烯纳米片在聚苯胺(PANI)中的分散情况.Fig. 9(a-d) Raman mapping of a GO sheet (a) and JGO (c) using theID/IG ratio from the corresponding Raman spectra (b, d) (Reproduced with permission from Ref.[ 21] Copyright (2020) Elsevier).3.3结晶度计算包括拉曼光谱在内的波谱技术经常被用于计算高分子晶体的结晶度. Mannanov等[23]利用原位拉曼光谱直接表征了应用于太阳能电池的P3HT:富勒烯基受体活性层中、P3HT在50~150 ℃温区的结晶动力学,并考察了溶剂、富勒烯基受体种类与结晶温度对P3HT结晶度的影响[14]. 结晶度的计算在选择合适的结晶特征峰与非晶特征峰基础上,结合光谱分峰/拟合处理及选择合适的结晶模型实现. 例如:Agarwal等[24]利用2种光谱分析方法计算了纤维素I晶体的结晶度,一种方法称为“单变量方法(univariate method)”,借助结晶峰/非晶峰强度的比值计算;另一种方法称为“多变量方法(multivariate method)”,应用偏最小二乘回归模型(partial least squares regression model)计算. 通过与已知结晶度的参比样品对比证实,2种方法在评价结晶度处于0%~80.5%范围内的纤维素样品时结果可靠,且由单变量方法得到的结晶度数值比由WAXS表征得到的更理想. Wang等[25]应用针尖增强拉曼光谱技术结合随机生长结晶模型,估算了合成的二维聚合物单层的结晶度,据此揭示了二维聚合物单层生长的交联本质[26].3.4分子链取向由于激光本身具有偏振性,如果使用偏光片对入射激光以及散射光的偏振方向进行调制,则可以获得高分子链中分子基团的取向信息,进而解析高分子链的取向结构,这种方法称为偏振拉曼[26~28]. 例如Richard-Lacroix等[26]使用偏振拉曼手段对使用不同收丝方法所得的静电纺聚氧化乙烯(PEO)单根纤维中PEO分子链的取向情况进行了研究. 测试过程中对于每一根纤维均需测试4组不同偏振角度的入射光与散射光的组合拉曼光谱,例如假设平行纤维轴方向为Z轴,垂直于纤维轴方向为X轴(Y轴暂不考虑),那么4组拉曼光谱分别为(X(入射光偏振方向)X(散射光偏振方向))、(XZ)、(ZX)与(ZZ). 不同的偏振组合所得的拉曼光谱中峰的强度有较大差别,说明分子链有取向存在,利用这些数据再通过进一步的计算便可以得出分子链的取向分布方程(orientation distribution function). Richard-Lacroix等的研究结果表明,单根纤维中的分子链总具有较高的取向并且与收丝方法无关.近年来,新的偏振拉曼数据分析手段也在不断地涌现,例如Richard-Lacroix等[28]提出了最可几分布(most probable distribution, MPD)方法,用以更加精确地定量分析分子链取向. Papkov等[29]利用一种改进的偏振拉曼分析方法,对直径分布在140~1000 nm范围的单根聚丙烯腈电纺纳米纤维的分子链取向进行了定量研究. Svenningsson等[30]基于包绕洛伦兹函数(wrapped Lorentzian function),开发了一种新的偏振拉曼光谱分析方法,并应用于确定再生纤维素纤维的分子取向研究. 这种方法的优势在于消除了偏振拉曼测试时对偏振角度的限制,所得结果能够与广角X-射线衍射与固体核磁的数据直接比较. 测量散射光偏振度随偏光片旋转角度的变化可以提供取向分布函数形状的半定性信息,Park等[31]据此分析了聚乳酸(PLLA)薄膜内部特征振动散射强度的角度依赖性,对结构单元的取向性进行了量化.3.5外场作用下的结构转变研究借助原位拉曼表征技术,能够对诸如温度变化[32~34]、时间改变[35]、拉伸过程[36,37]等的高分子结构演变进行追踪. Jin等[32]利用变温拉曼考察了高密度聚乙烯(HDPE)多重熔融行为中的构象变化. 作者对与熔融相关的变温拉曼光谱进行了如图10(a, b)所示的二维相干光谱分析(least squares moving-window method, LSMW),通过整个熔融过程中构象变化的相似性结合与“熔融-再结晶”、“中间相预熔融”及“多层片晶熔融”模型的比对,提出了如图10(c)所示的HDPE熔融时晶相直接转变为非晶相的机理. Kasiouli等[33]研究了β-环糊精包封的聚(4,4' -二苯基乙烯基) (PDV.Li)构象随温度的变化. 特征拉曼振动的强度变化证实,包封前后PDV.Li的主链平面性没有变化. 更高温度下主链构象的改变归因于由热诱导聚集引发的相邻苯环之间的扭转角度降低.Fig. 10(a,b) Least squares moving-window (LSMW) analysis of the HDPE Raman spectra with the window size of 11 spectra (ΔT = 1 °C). (a) A contour map of the first order derivative (d I/dT) as a function of Tave of a moving window. (b) The dI/dT of six Raman peaks are plotted after numerically integrated over frequency ranges to cover the Raman peaks: 1415-1425, 1115-1136, and 1299-1325 cm -1. (c) The schematic of the multithickness lamellae model. (Reprinted with permission from Ref.[32] Copyright (2017) American Chemical Society).基于拉曼光谱的多技术联用能够实现拉伸过程中高分子结构变化的表征与分析. Lόpez-Barrόn等[36]利用原位偏振拉曼技术,考察了线性低密度聚乙烯(LLDPE)拉伸过程中的单链构象及分子链取向变化. 结果表明,反式构象随拉伸程度的增加呈线性增加,分子链的伸展分为3个阶段,即弹性伸展阶段、塑性伸展阶段与应变硬化阶段. 取向因子受分子量影响,低分子量部分取向因子小. Kida等[37]利用原位偏振拉曼光谱与原位拉伸测试联用,考察了分子量分布对单轴拉伸过程中高密度聚乙烯形貌及变形行为的影响. 结果表明,连接片晶的带分子(tie molecules)数量随分子量分布的增大而增加,而晶体结构不受分子量分布影响. 晶区分子链沿拉伸方向的取向程度及连续的反式构象链的形成均在高分子量分布的样品中得到提高.3.6化学/物理组成研究表面增强拉曼光谱是一种能够在高分子共混结构的组分研究中提供潜在选择性与垂直分辨的强大技术. Razzell-Hollis等[38]借助此光谱探索了P3HT:聚((9,9-二辛基芴)-2,7-二基-alt-[4,7-双(3-己基噻吩-5-基)-2,1,3-苯并噻唑]-2' ,2"-二基)(F8TBT)共混薄膜的界面组成与分子有序性. 作者首先分别表征了P3HT与F8TBT的光谱,识别了由于样品退火引起的、与本体/界面形貌相关的光谱变化. 随后为了确定共混薄膜的化学组成,表征了不同共混样品的光谱并对光谱进行了分峰处理,获得了代表P3HT含量的强度值α与代表F8TBT含量的强度值β,结果见图11. 光谱分析的结果表明热退火改变了共混体系的界面组成:预退火增加了低表面能P3HT的含量,而后退火增加了高表面能F8TBT的含量. 此外,表面增强拉曼光谱还成功地应用于对纳米厚度尺度上高分子薄膜表面与底面的化学组分识别[39].Fig. 11Raman (a) and SERS (b) spectra for an as-cast sample of quartz (quartz) Q/Ag/P3HT:F8TBT, fitted using RR-P3HT (as ordered fraction), RRa-P3HT (as disordered fraction) and F8TBT spectra to obtain relative contributions of P3HT (α) and F8TBT (β). Normalized Raman (c) and SERS (d) spectra for P3HT:F8TBT blends in five different sample configurations, with variation in the relative intensity of the F8TBT peak at 1356 cm-1 shown in each inset (Reprinted with permission from Ref.[38] Copyright (2016) American Chemical Society).共聚焦显微拉曼技术近几年被广泛地应用于高分子多组分体系的化学/物理组成研究. 化学组分识别的相关研究涉及药物输送体系中聚乳酸-羟基乙酸共聚物[40]、聚己内酯和聚环氧乙烷的复合电纺纤维[41]、聚二甲基丙烯酰胺-甲基丙烯酸二苯甲酮共聚物[42]等. 物理组分识别方面,Hu等[43]研究了左旋聚乳酸/右旋聚乳酸(PLLA/PDLA)共混物球晶的等温结晶行为. 在如图12(a)和12(b)所示的800~600 cm-1波数范围内分别选择736与754 cm-1峰作为均晶与立构复合晶的特征峰,通过对球晶内部与外部两峰强度的成像分析(见图12(c)和12(d),证实大球晶内部包含均晶与立构复合晶2种晶体,立构复合晶均匀地分散在非晶区与球晶区域.Fig. 12Peak fitting results in the 800-600 cm-1 region at the single point at the position of 1# and 2# (a) and the peak fitting spectra of PLA with different crystal forms (b) Imaging result with imaging parameter: band intensity at 754 cm -1 (c) band intensity at 736 cm -1 (d) (Reproduced with permission from Ref.[ 43] Copyright (2019) Elsevier).其他成像方式如共振拉曼光电流成像(resonance Raman-photocurrent imaging, RRPI)[44,45]、飞秒激发拉曼成像(femtosecond stimulated Raman microscopy, FSRM)[46]、针尖增强拉曼成像(tip-enhanced Raman mapping, TERM)[47]、宽带相干反斯托克斯拉曼散射(broadband coherent anti-stokes Raman scattering, CARS)显微镜[48]、反转显微拉曼光谱(inverse micro-Raman spectroscopy, IMRS)[49]、等离子体波导共振拉曼光谱(plasmon waveguide resonance Raman spectroscopy, PWRRS)[50]等也应用于高分子化学组成分析.扫描角度拉曼光谱适用于分子有序程度的研究,能够同时获取增强的拉曼信号、薄膜厚度及分子有序程度的信息,此外结合均方电场计算(MSEF)可以确定聚合物薄膜中是否产生拉曼散射[51,52]. Meyer等[51]利用此光谱(示意图见图13(a))研究了P3HT:PCBM共混物在蓝宝石、金和铟锡氧化物界面处的形貌,考察了P3HT结构有序程度对基底的依赖性. 选择性激光入射角度下薄膜在蓝宝石基底上的拉曼光谱如图13(a)所示. 扫描角度从35°增加到60°,P3HT膜的拉曼强度呈现下降趋势,而当扫描角度进一步增加时拉曼强度提高. 与之不同,共混薄膜的拉曼强度随扫描角度的增加而持续下降. MSEF计算(见图13(b))揭示了拉曼信号在z方向上的距离依赖性,用于拉曼光谱的辅助解析,预期的拉曼信号与整个聚合物厚度上的积分MSEF成正比,这与实验的拉曼光谱一致. 此外,研究表明噻吩环C=C伸缩振动峰的宽度对P3HT的分子有序程度敏感,据此作者考察了分子有序程度对基底的依赖性.Fig. 13(a) Schematic of the SA Raman interface used to collect the data shown in B and C (A). SA Raman spectra at the indicated incident angles for (B) P3HT and (C) 1:1 P3HT:PCBM deposited on a sapphire substrate. (b) Calculated MSEF as a function of distance and incident angle for the interface: 0-1000 nm sapphire/1000-1230 nm P3HT:PCBM/1230-6000 nm air (A), 0-1000 nm sapphire/1000 to 1300 nm P3HT/1300 to 6000 nm air (B). The MSEF in the sapphire layer (0-1000 nm) and the majority of the air layer (greater than 1500 nm) are omitted for clarity. The calculated plots show the expected distance dependence of the experimental Raman signal in theZ direction. (Reprinted with permission from Ref.[ 51] Copyright (2013) American Chemical Society).4拉曼光谱应用展望激光拉曼光谱虽与红外光谱同属于分子振动光谱,但其拥有诸多红外光谱不可比拟的优势,例如高的空间分辨率、高解析度、测试范围横跨远红外与近红外光谱波段并且可以直接对水体系进行测试等. 如今伴随着新型高分子材料的不断涌现与应用,诸如高分子水凝胶,高分子纳米或多层复合材料等,以及表面增强拉曼,针尖增强拉曼以及共聚焦拉曼成像等新技术的接连出现,必将会使拉曼光谱在高分子材料的研究领域中迸发出强大的活力.然而与此同时,仍有一些问题限制了拉曼光谱的应用,例如在拉曼成像中,样品表面的高空间分辨率可以实现,但是垂直于入射激光深度方向上的空间分辨率则不佳,虽有研究使用金属粒子包埋在高分子样品中,再借助表面增强拉曼技术以实现高深度方向分辨率,但是这种方法的普适性稍显不足. 另外,如今拉曼成像技术一般仍为逐点扫描(mapping)模式,而红外成像则已多采用阵列扫描(imaging)模式,这就意味着拉曼成像需要较长的时间,从而很难使用拉曼成像进行过程研究,这也严重影响了拉曼成像的应用. 现今高分子的研究中多设备同步协同测试是一个趋势,例如X射线散射、拉曼及红外光谱同步在线测试,这也对拉曼设备的小型化以及快速响应提出了更高的要求. 相信通过拉曼设备以及技术的不断升级,这些问题都会迎刃而解,彼时拉曼光谱技术将会在高分研究领域占有更加举足轻重的地位.参考文献1Zhang Shulin(张树霖).Raman Spectroscopy with Low Dimensional Nanometer Semiconductors(拉曼光谱学与低维纳米半导体).Beijing(北京):Science Press(科学出版社),2008.3-352Koenig J L.Spectroscopy of Polymers.Netherlands:Elsevier,1999.207-252.doi:10.1016/b978-044410031-3/50005-03Chalmers J,Griffiths P.Handbook of Vibrational Spectroscopy, 5 volumes set.New Jersey:John Wiley & Sons,2002.1-174Sasic S,Ozaki Y. Raman,Infrared, andNear-Infrared Chemical Imaging.New Jersey: John Wiley & Sons,2011.1-215Schrader B.Infrared and Raman Spectroscopy: Methods and Applications.New Jersey:John Wiley & Sons,2008.7-616McCreery R L.Raman Spectroscopy for Chemical Analysis.New Jersey:John Wiley & Sons,2000.15-30.doi:10.1002/04717216467Colthup N B,Daly L H,Wiberley S E.J Am Chem Soc,1965,87(5):1155-11568Wilson E B,Decius J C,Cross P C,Sundheim B R.J Electrochem Soc,1955,102(9):235C.doi:10.1149/1.24301349Tadokoro H.Structure of Crystalline Polymers.New Jersey:John Wiley & Sons,1979.179-322.doi:10.1002/macp.1979.02002197911010Larkin P.Infrared and Raman Spectroscopy.Netherlands:Elsevier,2011.7-25.doi:10.1016/b978-0-12-386984-5.10002-311Dieing T,Hollricher O,Toporski J.Confocal Raman Microscopy.Berlin:Springer,201112Gautam R,Samuel A,Sil S,Chaturvedi D,Dutta A,Ariese F,Umapathy S.Curr Sci,2015:341-356.doi:10.1140/epjti/s40485-015-0018-613Gao J,Thomas A K,Johnson R,Guo H,Grey J K.Chem Mater,2014,26(15):4395-4404.doi:10.1021/cm501252y14Martin E,Bérubé N,Provencher F,Côté M,Silva C,Doorn S,Grey J.J Mater Chem C,2015,3(23):6058-6066.doi:10.1039/c5tc00847f15Yu W,Zhou J,Bragg A E.J Phys Chem Lett,2012,3(10):1321-1328.doi:10.1021/jz300329816Gao Y,Grey J K.J Am Chem Soc,2009,131(28):9654-9662.doi:10.1021/ja900636z17Gao Y,Martin T P,Thomas A K,Grey J K.J Phys Chem Lett,2010,1(1):178-182.doi:10.1021/jz900038c18Gao J,Grey J K.J Chem Phys,2013,139(4):490319Gao J,Thomas A,Yang J,Aldaz C,Yang G,Qin Y,Grey J.J Phys Chem C,2015,119(16):8980-8990.doi:10.1021/acs.jpcc.5b0216620Wang M,Vantasin S,Wang J,Sato H,Zhang J,Ozaki Y.Macromolecules,2017,50(8):3377-3387.doi:10.1021/acs.macromol.7b0013921Zhang Z , Qin J , Diao H , Huang S,Yin J,Zhang H,Duan Y,Zhang J.Carbon,2020,161:316-322.doi:10.1016/j.carbon.2020.01.07822Badi N,Khasim S,Roy A S.J Mater Sci Mater Electron,2016,27(6):6249-6257.doi:10.1007/s10854-016-4556-823Mannanov A A,Bruevich V V,Feldman E V,Trukhanov V A,Pshenichnikov M S,Paraschuk D Y.J Phys Chem C,2018,122(34):19289-19297.doi:10.1021/acs.jpcc.8b0313624Agarwal U P,Reiner R S,Ralph S A.Cellulose,2010,17(4):721-733.doi:10.1007/s10570-010-9420-z25Wang W,Shao F,Kroger M,Zenobi R,Schluter A D.J Am Chem Soc,2019,141(25):9867-9871.doi:10.1021/jacs.9b0176526Richard-Lacroix M,Pellerin C.Vib Spectrosc,2017,91:92-98.doi:10.1016/j.vibspec.2016.09.00227Richard-Lacroix M,Pellerin C.Macromolecules,2012,45(4):1946-1953.doi:10.1021/ma202749d28Richard-Lacroix M,Pellerin C.Macromolecules,2013,46(14):5561-5569.doi:10.1021/ma400955u29Papkov D,Pellerin C,Dzenis Y A.Macromolecules,2018,51(21):8746-8751.doi:10.1021/acs.macromol.8b0186930Svenningsson L,Lin Y C,Karlsson M,Martinelli A,Nordstierna L.Macromolecules,2019,52(10):3918-3924.doi:10.1021/acs.macromol.9b0052031Park M,Wong Y S,Park J,Venkatraman S,Srinivasarao M.Macromolecules,2011,44(7):2120-2131.doi:10.1021/ma101553v32Jin Y,Kotula A P,Snyder C R,Hight Walker A R,Migler K B,Lee Y J.Macromolecules,2017,50(16):6174-6183.doi:10.1021/acs.macromol.7b0105533Kasiouli S,Di Stasio F,McDonnell S O,Constantinides C P,Anderson H L,Cacialli F,Hayes S C.J Phys Chem B,2013,117(18):5737-5747.doi:10.1021/jp400732h34Winfield J M,Donley C L,Friend R H,Kim J S.J Appl Phys,2010,107(2):1073.doi:10.1063/1.327625735Magnanelli T J,Bragg A E.J Phys Chem Lett,2015,6(3):438-445.doi:10.1021/jz502605j36López-Barrón C R,Zeng Y,Schaefer J J,Eberle A P R,Lodge T P,Bates F S.Macromolecules,2017,50(9):3627-3636.doi:10.1021/acs.macromol.7b0050437Kida T,Hiejima Y,Nitta K.Macromolecules,2019,52(12):4590-4600.doi:10.1021/acs.macromol.8b0274038Razzell-Hollis J,Thiburce Q,Tsoi W C,Kim J S.ACS Appl Mater Interfaces,2016,8(45):31469-31481.doi:10.1021/acsami.6b1212439Linde S,Carella A,Shikler R.Macromolecules,2012,45(3):1476-1482.doi:10.1021/ma201867e40McManamon C,Delaney P,Kavanagh C,Wang J J,Rasappa S,Morris M A.Langmuir,2013,29(19):5905-5910.doi:10.1021/la400402a41Kotzianova A,Rebicek J,Mojzes P,Pokorny M,Palacky J,Hrbac J.PolymerVelebny V,2014,55(20):5036-5042.doi:10.1016/j.polymer.2014.08.03242Janko M,Jocher M,Boehm A,Babel L,Bump S,Biesalski M,Meckel T,Stark R W.Biomacromolecules,2015,16(7):2179-2187.doi:10.1021/acs.biomac.5b0056543Hu J,Wang J,Wang M,Ozaki Y,Sato H,Zhang J.Polymer,2019,172:1-6.doi:10.1016/j.polymer.2019.03.04944Gao Y,Martin T P,Thomas A K,Grey J K.J Phys Chem Lett,2010,1(1):178-182.doi:10.1021/jz900038c45Grey J K.Acc Chem Res,2019,52(8):2221-2231.doi:10.1021/acs.accounts.9b0008846Nixdorf J,Di Florio G,Bröckers L,Borbeck C,Hermes H E,Egelhaaf S U,Gilch P.Macromolecules,2019,52(13):4997-5005.doi:10.1021/acs.macromol.9b0020547Xue L,Li W,Hoffmann G G,Goossens J G P,Loos J,de With G.Macromolecules,2011,44(8):2852-2858.doi:10.1021/ma101651r48Lee Y J,Snyder C R,Forster A M,Cicerone M T,Wu W L.ACS Macro Lett,2012,1(11):1347-1351.doi:10.1021/mz300546e49Raupp S M,Siebel D K,Kitz P G,Scharfer P,Schabel W.Macromolecules,2017,50(17):6819-6828.doi:10.1021/acs.macromol.7b0103750Meyer M,McKee K,Nguyen V H T,Smith E.J Phys Chem C,2012,116(47):24987-24992.doi:10.1021/jp308882w51Meyer M W,Larson K L,Mahadevapuram R C,Lesoine M D,Carr J A,Chaudhary S,Smith E A.ACS Appl Mater Interfaces,2013,5(17):8686-8693.doi:10.1021/am402322552James D T,Kjellander B K C,Smaal W T T,Gelinck G H,Combe C,McCulloch I,Wilson R,Burroughes J H,Bradley D D C,Kim J.ACS Nano,2011,5(12):9824-9835.doi:10.1021/nn203397m原文链接:http://www.gfzxb.org/thesisDetails#10.11777/j.issn1000-3304.2020.20251&lang=zh《高分子学报》高分子表征技术专题链接:http://www.gfzxb.org/article/doi/10.11777/j.issn1000-3304DOI:10.11777/j.issn1000-3304.2020.20251






















 我要推广仪器
我要推广仪器
 下载APP
下载APP