[color=#444444]我的样品分子量大约是700,在计算时候却算成了300,质谱谱图回来时候看见扫描范围仅到500. [/color][color=#444444]问:做质谱的老师会不会因为没扫那个范围而把分子离子峰屏蔽了?还是在我要的700处根本就没有那个分子离子峰?做质谱时候扫描范围都定在多少区间?[/color]
初学者,请问质谱扫描范围可以从m/z=0开始吗
质谱中超过了扫描范围还会出峰吗? 我的质谱有明显的峰,但是不确定具体分子量,就选了100-2000进行[url=https://insevent.instrument.com.cn/t/Yp][color=#3333ff]液质联用[/color][/url]的scan,正模式有明显的峰但是离子很干净只有274和318 (推测是杂质),我的目标化合物的分子量很大,就想问问是不是因为超过了扫描范围,看不见离子对?
[color=#333333]请问质谱扫描范围可以从m/z=0开始吗[/color]
质谱扫描某种特定的离子(如M/Z=181)时,为什么它的特征峰是一个范围呢?比如说我选择定性离子181,它的选择离子峰是一个范围,会有181.1~181.x呢
http://ng1.17img.cn/bbsfiles/images/2012/03/201203101047_353720_2359621_3.jpg质谱灵敏度降低能否改变扫描速度和质量范围提高
我们在使用质谱仪时,常常会用到一个名词-扫描速度。顾名思义,扫描速度就是指的扫描M/Z的速度,即1秒内,可以扫描M/Z的范围。 如果扫描速度是1000,则表示1S内,可以扫描1000M/Z。 扫描速度跟扫描范围和扫描周期有关。如果扫描范围是100M/Z,而扫描周期是100ms,则理论上扫描速度是1000/100*100=1000M/Z。这里第一个1000指的是1000ms。上面说到这只是理论的扫描速度,而我们使用的质谱软件计算出来的扫描速度往往不是1000,这是为什么呢?这就要跟实际的硬件结构挂钩了。实际上,仪器在做周期扫描时,每个周期之间会有一段平衡时间,根据不同的仪器,平衡时间是不一样的。假如以3ms为例。则实际上100ms里只有97ms是可以进行扫描的。所以:100/(100-3)*1000=1030.92 ,这里第一个100是扫描范围,第二个100是扫描周期,单位ms,1000是扫描速度,这里是固定值,做基准。 上面已经计算出理论扫描速度是1030,所以1/1030*1000*1000=97.087。这里计算出的97.08是理论上DC扫描的间隔时间,而实际应用中,DC扫描的间隔时间会把各位去除,取十位和十位以上的整数部分。所以DC间隔时间为90us。1*1000*1000/90=11111。所以1秒内DC一共输出11111次,而DC每输出一次,扫描0.1M/Z,所以扫描速度为11111*0.1=1111。所以我们可以看到工作站软件计算出来的扫描速度是1111。
假如您分析采集的质谱数据的扫描范围不对,例如扫描起点太高,一部分低的离子没有扫到,或者高端的离子或者分子离子没有扫到,请问您会怎样办?
对于逐点扫描得到的一段质谱数据,数据处理的首要任务是峰位置的判别。其实质是峰数据与既有模型的匹配过程,这与质谱仪的特性、扫描参数以及数据的统计信息等多种因素有关系。简单情况下,连续几个数据都大于设定的阈值(如最大值5%)即可认为该段数据是峰数据,而剩余的数据可认为是本底。在峰位置判别的基础上,根据本底数据判断谱段的基线。可将感兴趣谱段的非峰数据(未被标记)的平均值作为基线。但对于大范围的质谱扫描谱,可能存在不同谱段本底不同的现象,因此当处理几十个质量扫描范围质谱数据时,应注意基线的波动。对于每个具有一定幅度的质量峰,确定其峰中心位置是数据处理的重要一环。质量峰的位置准确,才能正确地反映离子流强度的变化。对于左右对称的峰,其峰中心一般取两个半高横坐标的中心;对于左右不对称的峰,可分别对峰两侧的斜坡作延长线,两延长线的交点位置即可作为峰中心。在作峰中心时,数据的涨落往往给计算结果带来显著的偏差,这也是峰中心标定的误差来源。对于平顶不明显的谱图,可以使用二次曲线拟合得到离子流强度。对于每个峰位置,原始数据的横坐标可能是计算机设定的DAC数值,也可能是按照时间排列的序列数。要通过计算机自动标定每个峰位置对应的质量数,除了要求一定的峰数据的量,还必须有对应的扫描参数和数据库支持。可人工指定几个峰位置对应的质量数,再由计算机根据扫描参数与质量数之间的线性或非线性关系算出其他相邻峰的位置,从而可画出峰强度质量谱图。对扫描峰离子信号的强度计算,第一种是峰高法,用峰中心位置的数据(或连续几个数据的均值)减去基线数据作为离子信号强度;第二种是峰面积法,用该峰数据(一般选大于5%峰高的数据)和基线围成的面积作为离子信号强度;第三种是采用窗口数据累加,即以峰中心位置开始向大质量数和小质量数寻找固定长度,确定一个质量范围,将该质量范围内的数据平均值减去基线数据作为离子信号强度。离子峰数据的涨落和基线的涨落都对测试数据有较大的影响,比较而言,峰面积法的精度高于其他方法。通过对峰数据的分析,还可得到其他质量峰的特征参数:①半峰宽。是反映仪器分辨本领的参数之一。谱图在一半峰高处的质量数之差就是半峰宽。②峰顶平坦度。反映探测器的稳定度。只有梯形峰谱图才能计算,计算公式为平顶位置处的离子流强度的极差与峰高的比值。该值越小表明探测器越稳定。③峰形系数。是反映仪器分辨本领的参数之一。定义为10%峰高处的峰宽与90%峰高处的峰宽之差与峰半高全宽的比值,该值用百分比表示。
1. 阻抗谱测试,参数设定的起始电位是?2. 我的线性扫描电势得到的E-I曲线,怎么全部是线性区? (可以保证扫描范围已经很大)3. 电势阶跃(计时电流)法参数设定的起始电位是?脉冲宽度设定?4. 循环伏安的扫描范围是?比如研究溶液对工作电极的氧化等作用, 扫描起始电位应该高于开环电势还是怎么设定?多谢!!!
各位好:求助拉曼光谱选择扫描范围和激发波长 急!!!我作了个样,用拉曼光谱表征,物质为硅胶负载有机物(对甲苯磺酸盐类),但好像荧光比较明显,干扰大,检测老师叫我提供扫描范围和激发波长,不是太懂,真的很急,请各位虫虫帮忙 谢谢!!!
[font=&][color=#333333]质谱仪扫描方式有两种:全扫描和选择离子扫描[/color][/font][font=&][color=#333333]。全扫描是对指定质量范围内的离子全部扫描并记录,得到的是正常的质谱图,这种质谱图可以提供未知物的分子量和结构信息。可以进行库检索 。质谱仪还有另外一种扫描方式叫选择离子监测( Select bn Mo[url=https://insevent.instrument.com.cn/t/1p][color=#3333ff]NIR[/color][/url]ing,SM)。这种扫描方式是只对选定的离子进行检测,而其它离子不被记录。它的最大优点是对离子进行选择性检测 、只记录特征的 、感兴趣的离子,不相关的、干扰离子统统被排除;是选定离子的检测灵敏度大大提高,采用选择离子扫描方式比正常扫描方式灵敏度可提高大约 100 倍。由于选择离子扫描只能检测有限的几个离子,不能得到完整的质谱图,因此不能用来进行未知物定性分析。但是如果选定的离子有很好的特征性,也可以用来表示某种化合物的存在。选择离子扫描方式最主要的用途是定量分析,由于它的选择性好,可以把由全扫描方式得到的非常复杂的总[url=https://insevent.instrument.com.cn/t/3p][color=#3333ff]离子色谱[/color][/url]图变得十分简单,消除了其它组分造成的干扰。在一般色谱分析中主峰对被测组分的影响很大,为了降低主峰的影响,通常采用预切割技术使得仪器的气路比较复杂,操作比较麻烦 。而质谱的优点就是通过离子选择性技术很方便地避开了主体组分的影响 。[/color][/font]
各位: 大家好!我想问一下扫描电镜能谱所能分析的最小范围是多大。 谢谢各位!
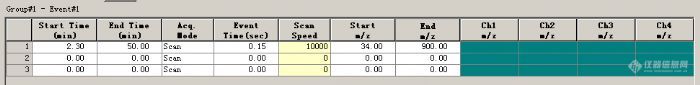
各位老师,第一张图我划线的部分,扫描范围是第二张图m/z的扫描范围吗,个人感觉不对呀,我也没找到有设置扫描范围的地方,第二个问题是第二个图的扫描速度根据啥因素设置,昨天看着资料说设置的好出现的峰好[img]https://ng1.17img.cn/bbsfiles/images/2019/09/201909291023210103_5359_4004805_3.png[/img][img]https://ng1.17img.cn/bbsfiles/images/2019/09/201909291023210393_393_4004805_3.png[/img]
全扫描采集模式,扫描范围为29-350,但出现的峰中第一个总是会出现氧气峰,而且峰面积很大。不清楚这个峰哪儿来的?是不是应该把扫描范围的最小值调高点。谢谢了
请问测试三维谱图时,激发波长扫描范围和发射波长扫描范围怎么设置,另外Em1 start offset如何设置? 如有回复,不胜感激!!

(转载引用请注明出处)质谱计扫描速度是什么? 这是安捷伦公司提供的一幅两种质谱检测结果表示方法的对照和说明:http://ng1.17img.cn/bbsfiles/images/2017/10/2015033009161697_01_2991869_3.jpgQuadrupole DescriptionProfile versus spectrum scansIn the upper plot, a profile scan of an ion at m/z 109 is shown. In a typical benchtop mass spectrometer, abundance measurements are collected at 0.10 m/z increments as shown.When this data is presented in a mass spectrum, a single line is shown. The height and position are derived from the profile scan. Notice that in the plot of the spectrum, a single line is used to indicate that a single mass is present.译文: 四级杆简介 峰截面轮廓曲线图与与之相对应的各质谱图数据子集扫描 在上面的图中, 展示了对m/z 109离子的扫描结果。 在标准的台面型质谱计中, 丰度测得值是以0.1 0 m/z为单位步进收集的。 当这一数据集以质量谱图的形式表达时, 我们只能看到一根直线。 直线的高度和位置来自对峰截面轮廓线式扫描结果的解释。 注意在谱线式图形中, 只用了一根直线表示只存在一种质量数。 这段文字说明了什么意思呢? 说明了一般所讲的质谱计扫描速度, 指的是“驱动不同质量离子的动力变化速度”。 什么东西能以“0.1m/z”为单位步进呢? 击中探测器的离子数不行, 离子的质量数都大于1。 在高分辨质谱图中最少也是一点几几几, 在低分辨率质谱图中就是1(氢)、 2(氦)、 ... ... 、 12(碳), 等等之类。 探测器也只会记录电脉冲数, 分不出来离子质量。 “机器的检测时间(莫专家语)”不行, 时间的单位是时、分、 秒、 毫秒(ms)、 纳秒(ns)等等之类。 只有使不同质量的离子分开的加在四级杆和离子阱上的电压可以。 电压是可以“步进”并被精准记录的, 可电压的单位是伏特(V), 那么, 只有将可以只驱动0.1m/z质量的微粒进入四极杆飞向探测器, 或者只可以将0.1m/z质量的微粒驱出离子阱飞向探测器的电压伏特数, 在质谱计中可能是“纳伏数”, 计算转换表达为m/z值, 就有了“以0.1 0m/z为单位步进收集”的“丰度测得值”。 为什么上面的“峰截面轮廓线图”会变成下面的“直线图”呢? 这是因为在电压以0.1m/z步进到到109m/z之前, 因为没有107.99、108.00、 108.10、 108.20的离子, 高分辨力质谱计的高灵敏探测器能测到108.***的离子和109.***的离子, 低分辨力质谱计的探测器啥也测不到, 所以这时没有“丰度”被探测器收集到; 在109m/z之后, 同样没有109.10、 109.20、 ... ...、 110.00、110.10之类的离子, 探测器又啥都收集不到了, 只有在电压步进到109.00m/z时, 探测器才能收集到m/z 109的离子, 在低分辨力质谱计中将109.00左边的108.5**以上的离子和右边的109.500以下的离子(大致是这样吧)统统归入这个m/z109离子的计数值(丰度), 再将其与质量数对应起来, 就是下面的“直线图”或曰“柱状图”。 我们知道, 质谱计的探测器只能记录电脉冲数,1amu的氢离子是一个脉冲, 几千amu的蛋白质分子也是一个电脉冲, 如果只从探测器的结果记录, 则只有时间-电脉冲数记录, 而这些电脉冲是多大质量的离子产生的, 则无法分辨。 那么只有让一种质量的离子在一个时间段到达探测器, 才能分辨出来时间序列上的每组计数的离子质量(m/z值)。 用什么办法使不同质量的离子在一个时间段有序到达探测器呢, 只有改变施加在四级杆和离子阱上的控制电压, 射频(RF)或者直流(DC), 才能做到, 而电压的变化是能被仪器精确记录的, 精确记录的电压与粒子质量是精确对应的, 所以就可以将由小到大变化的, 驱动质量由小大的离子顺序飞向探测器的电压变化速度表达为单位时间内质量数的变化率, 也就是质谱计的以amu/秒为单位的“扫描速度”, 质谱计的5600amu/sec的扫描速度的意思, 就是仪器可以在一秒钟之内顺序将质量数从1amu到5600amu的离子顺序驱离出阱或者分质量过杆, 只是这个扫描速度是驱动离子分质量“过杆”或者“弹出离子阱”的电压变化速度, 不是探测器实际测到的脉冲数。 安捷伦公司的文章作者和译员喜欢将“驱动离子出阱”写成“扫描离子出阱”, 在这里, 我不愿意用“scan”这个词。
质谱仪作为目前较常用的对纯物质鉴定的工具,在生产生活中具有着重要的地位。一般常见的质谱仪具有全扫描、单离子监测、选择离子扫描、中性碎片丢失扫描等。以下收集了一些质谱扫描模式相关信息,以便更具体了解质谱: [b]1.全扫描 Full?Scan[/b] 全扫描是指在进行质谱采集时,扫描一段自己来设定的范围,然后覆盖对被测化合物的分子离子以及碎片离子的质量,一般想要得知未知物质的分子量以及相应的所有的碎片离子,都会进行全扫描,通过扫描得到的全谱,进行相应的谱库检索从而得到未知物质的相应信息。[b] 2.单离子监测 SIM[/b] 单离子监测也称选择离子扫描,是指针对一级质谱而言,进行只扫一个离子的模式,并不是连续扫描某一范围内的质量,而是通过设定有针对的扫描某几个选定的质量。一般对已知物质,为避免其他离子对实验的干扰并提高某一离子灵敏度,通常仅需要扫描一个离子。一般用于化合物检测和复杂混合物中杂质的定量扫描检测。 [b]3.中性碎片丢失扫描 Neutral Loss[/b] 在进行中性碎片丢失扫描时,质量分析器选择所有离子进入碰撞室,与碰撞室内的碰撞气体发生碰撞诱导裂解,另一个质量分析器以与前一个质量分析器固定质量差联动扫描,检测丢失该固定质量中性碎片的离子对,得到中性碎片质谱。 [b]4.选择反应监测 SRM[/b] 选择反应扫描也称作选择反应监测,是一种针针对二级质谱或多级质谱的某两级之间的扫描模式。当母离子选一个离子发生碰撞后,从形成的子离子中也只选一个离子。选择反应监测由于两次都只选单离子,因此能排除掉大部分的干扰信息,在相对复杂且基质背景高的样品检测中,其灵敏度信噪比会较高,抗背景排干扰的能力也较强。 [b]5.多反应扫描 MRM[/b] 多反应扫描既多反应监测,是在进行多个化合物检测过程中,多个选择反应监测一起进行。因此多反应扫描的特点与选择反应监测特点相似,因此在实际操作生产中并不会对选择反应监测与多反应监测进行区分,在实验过程中只要同时进行多个选择反应监测便是多反应监测了,彼此之间区分仅是数量上的分别。 在一般进行质谱实验的过程中,对于检测物质定性扫描时,因为想观测到更多的离子因此会选择全扫描模式。当定量检测时,一般会想要提高已知的信息强度在复杂的基质环境下的测量,会选择单离子监测、选择反应监测以及多反应监测等
请问您怎样您怎样确定全扫描scan)的质量范围?或您全扫描时候的质量范围是多少?有人讲最高质量是分子量M+50, 有人讲避开28,32等低质量离子的干扰,您认同吗?或您有什么好的想法来设置全扫描质量范围?
有个循环伏安特性曲线的问题向各位大侠请教,万分感谢各位的帮助:我测了循环伏安特性曲线来表征超级电容器(活性炭电极,KOH电解质)循环伏安特性,扫描范围是从0V到+1V,扫描速率为50mV/s。有人建议我扫描范围改为-1到+1V,不知道道理是什么,有何区别?再次感谢!
新人学习XRD,现在遇到了问题,比如说检测样品中是否含有石英,但是不知道石英会在哪个角度有峰值,不知道扫描范围设定在多少,求助一下哪位能教教我,感激不尽!
GCMS使用调查:在做全扫描时,您使用扫描范围是多少?为什么?
仪器参数如下(agilent6890-5973)1.质量范围:1.6~800u,2.精度0.1u 3.扫描速度:在0.1u步进速度下,最大可达5200u/s4.一般分析情况下,可选择速度800u/s 如果仪器在scan状态下采集数据,设定质量范围为50u-350u,扫描速度为900u/s(扫描速度不知道在工作站哪里设置,是不是用数据采集那里2的几次方有关?)这样设置参数的话,是不是就是说质谱在1s内,可以从50u扫描到350u扫描3次,也就是在一秒内可以得到3张质谱图?望各位高人指点,希望用这款仪器的同行告诉我扫描速度在工作站中的设定。中国心
假如全扫测定时候由于扫描范围不是很合适,只是采集到部分离子,请问您可能会用什么办法来检索呢?质谱图很难以检索例子2 [url]https://bbs.instrument.com.cn/topic/7459118[/url]
5000:1 这个扫描间动态范围 如何理解?
我刚开始做极谱分析,用的是江苏电化学仪器厂的产品,现有一问题想求教各位,如下: 如选择-0.4到-0.8的扫描范围那么,在-0.4前会产生峰的物质会不对采样产生干扰?谢谢!
我个人认为, 质谱计的扫描速度不要用800amu/s, 或5000amu/s来表示。 用800个离子/s、 5000个离子/s来表示更好。 否则易引起误解。 不知各大质谱制造公司是否认同?因为H+离子是一个电脉冲, 5000amu的离子也是一个电脉冲。
大家平时扫描范围是多少?尤其是起始数值,我之前用的都是45amu,避开了氮气,氧气,二氧化碳最近分析新样品,需要从20开始,发现有氮气,氧气,二氧化碳碎片的干扰,机器开机已经48小时了,这个问题应该怎么解决比较好?谢谢
请问做全扫时候,您的扫描范围是多少?那类样品?
关于这个扫描速度应该是每秒钟能扫描的质量数有多少吧?看到有些质谱的技术参数上写的是最大能达到多少的扫描速度,这个是和你所选择的质量范围相关的吗?







 我要推广仪器
我要推广仪器
 下载APP
下载APP