2003年人类基因组精细图绘制完成,是人类科学史上一个里程碑式的事件。后基因组时代的研究重点自然落在了蛋白质头上。为啥?因为中心法则告诉我们,基因的产物——蛋白质,是生命活动的最终执行者。与基因组类比,研究生物体内全套蛋白质的科学,就是蛋白质组学。基因组计划完成的同年,人类蛋白质组计划启动,令人激动的是,2014年人类蛋白质组的草图也完成了。而蛋白质组学能够飞速发展的最大功臣非质谱莫属。质谱的应用范围非常广泛,但这里只讨论蛋白质组学中的质谱。简单地说,质谱法(mass spectrometry)就是对肽段离子的重量(质荷比,m/z)进行测量的分析方法。样品经质谱仪(mass spectrometer)检测得到质谱图(mass spectrum),通过对质谱图的分析就可以对样品中的蛋白进行鉴定、定量。亲,图1的这种典型的蛋白质组学流程都很熟悉吧。蛋白首先都要被特异性的酶(通常为Trypsin)切割为肽段,再进行后续分析,这在蛋白质组学中被称为“自下而上”的研究策略(Bottom-up proteomics)。我们平时见到的质谱分析基本都是这种类型。提到蛋白质组,即会联想到一系列高大上的名词,iTRAQ、SWATH、SILAC、Shotgun、Label-free等等。很多概念容易弄混淆,下面我们就来理理清楚。图1. 典型的蛋白质组学流程大体上,质谱研究蛋白主要是鉴定和定量。通过二级质谱图(MS2或者MS/MS)进行数据库搜索匹配鉴定蛋白。通过各种标记或非标记的手段对不同样品中的蛋白进行比较就是定量。蛋白定量比较是质谱最重要的用途,图2是对定量方法的一个简单总结。非标定量(Label-free)不需要标记,不同样品分别处理、分别进质谱检测;优点是处理简单、无需标记、价格便宜、可以比较很多组样品,缺点是对操作步骤、LC、质谱稳定性要求严格。SILAC是在细胞培养基中加入稳定同位素标记的氨基酸,在代谢水平标记蛋白,一级质谱图进行定量,可以做到三组样品混合后进行比较,定量准确,但是不能标记组织样本,养细胞成本也较贵。双甲基化标记是通过化学反应的办法在肽段水平进行标记,一级质谱定量,也可以三组对比,标记试剂都比较便宜,而且可以标记任何来源的样品。iTRAQ和TMT是商品化的试剂盒,肽段水平标记,二级质谱定量;分别可以做到最多8组和10组样品间蛋白质组的比较。图2. 质谱定量方法以上这几个是一家的,还有几个名词是属于另外一家,比如Shotgun (DDA)、SWATH/DIA、SRM (MRM)、MRMHR/PRM。质谱进行数据采集的方式大致分为三种:鸟枪法(Shotgun)、选择反应监控(SRM)和全景式的SWATH/DIA。下面对照图3再来简单介绍一下。图3. 质谱扫描方式DDA、IDA、Shotgun和鸟枪法说的是相同的东西,意思是质谱在每个循环的中从一级里挑选丰度高的TopN个肽段去打碎做二级扫描,得到的结果通过与已知数据库中的理论蛋白进行匹配。DDA简单有效,分析流程比较成熟,也是目前质谱分析的主流方式。DDA也有其固有的缺陷,即具有一定的随机性,偏向于检测丰度较高的肽段,而抑制了低丰度肽段的检测。靶向策略被称为质谱领域的Western blot。质谱只去采集目标肽段大小的离子信息,因而提高了灵敏度和特异性。这种方法用来研究感兴趣的特定蛋白,定量准确,但是通量很有限。SWATH/DIA这种全景式的数据采集方式在最近几年突然火了起来,被认为在不远的未来可能会取代DDA的主流位置。该方法采取的策略是将扫描范围内的所有肽段按照质荷比分为若干个窗口,再对每个窗口里所有的肽段一起打碎,采二级,数据分析时通过抽提蛋白的子离子信息进行定量。SWATH/DIA解决了DDA中随机性选择肽段的缺陷,所以重复性更好,定量的准确性基本达到了SRM的水平,而且可以实现大规模定量。借用听来的一个比喻来说明:DDA就像机关枪扫射,数量多、体积大的目标命中的概率要大一些。靶向扫描(SRM或PRM)就像精准狙击,排除干扰,目标明确,每一枪直指目标,但是难以大规模消灭敌人。SWATH/DIA就是地毯式轰炸,只要暴露在我方攻击范围内的敌人,不管三七二十一,全部炸完。图4. 定量方法与采集方式结合如果将上述的定量方法(图2)和质谱数据采集方式(图3)结合起来,就得到了现在基于质谱的蛋白质组学研究的各种策略(图4)。再打个比方,保证吃货们一听就懂:鸡、鱼、肉、蛋、蔬菜要通过炒锅、烤箱、高压锅、微波炉等烹调之后才能变为美食,填饱肚子。同样的,各种定量方法(非标的和标记的)处理的样品,要通过质谱各种采集方式变为电脑中的数据,才能分析并从中得到蛋白的信息。本次的介绍就先到这里了,如果其中有什么问题,欢迎您批评和建议,我们会努力变得更好;如果需要跟我们进行技术交流和讨论,欢迎大家联系武汉金开瑞。后续我们还会继续推出对质谱技术各方面进行解析的文章,敬请期待。ReferencesA draft map of the human proteome. Nature 509: 575–581 (2014)Mass-spectrometry-based draft of the human proteome. Nature 509: 582–587 (2014)A review: Annu. Rev. Biochem. 80: 273–99 (2011)SILAC: Molecular & Cellular Proteomics 1: 376-386 (2002)iTRAQ: Molecular & Cellular Proteomics 343: 91–99 (2010)SRM: Nature Methods 9: 555–566 (2012)SWATH: Molecular & Cellular Proteomics 11: 1–17 (2012)
褚福亮,王福生, 中国人民解放军第302医院全军艾滋病与病毒性肝炎重点实验室 北京市 100039项目负责人 王福生, 100039 ,北京市丰台路26号, 中国人民解放军第302医院全军艾滋病与病毒性肝炎重点实验室. fswang@public.bta.net.cn电话:010-66933332 传真:010-63831870收稿日期 2002-08-15 接受日期 2002-09-03摘要新近广泛应用蛋白质芯片(ProteinChipâ Array)系统成功鉴定出了一些重要疾病(如肿瘤和危害性较大的传染病)新的、特异性的生物标记(biomarkers),后者不仅在生物医学的基础方面具有重要的科学价值,而且在临床疾病的诊断、治疗和预防发挥重要的指导作用,显示了良好的发展前景.本文就表面增强的激光解析电离-飞行时间-质谱(SELDI-TOF-MS)相关的原理、特点、在临床和基础研究中的应用新进展和未来的发展趋势做一综述.此外,我们就蛋白质谱分析技术在病毒性肝炎、肝硬化和肝癌等一系列肝病方面的应用策略和前景进行了分析.褚福亮,王福生. 蛋白质谱分析方法特点及其在蛋白组学研究领域中的应用.世界华人消化杂志 2002 10(12):1431-14350 引言人类基因组计划已经进入后基因组时代-即功能基因组时代[1],作为基因功能的直接体现者-蛋白质,及其之间的相互作用越来越引起基础和临床科学家们的关注[2-6] .因为要彻底了解生命的本质,只把基因测出来还是不够的,还必须要了解其在生物生长、发育、衰老和整个生命过程中的功能、不同蛋白质之间的相互作用以及他们与疾病发生、发展和转化的规律[7-14] .正因为如此,有关上述问题的蛋白质组学研究成了今天生命科学最重要的焦点之一[15] .为了阐明蛋白质在上述生命现象中的作用和相关机制,人们设计了许多新的方法技术,如:二维电泳、质谱分析、微距阵列、酵母双杂交和噬菌体展示等,这些方法在一些特定的情况下,虽然显示出了他们各自不同的优点,但是同样也存在着较大的局限性,难以开展大规模、超微量、高通量、全自动筛选蛋白质等方面的分析,因而设计更全面、同时研究多种蛋白质相互作用的技术,在功能基因组和蛋白组学的研究中建立一个更有效的技术平台,成为本领域中优先关注的问题[16] .近来,美国Ciphergen(赛弗吉)公司研制的ProteinChipâ Array的仪器,并建立了一种新的蛋白质飞行质谱-表面增强的激光解析离子化-飞行时间-质谱(surface-enhanced laser desorption/inionation-time of flight-mass spectra, SELDI-TOF-MS),已取得可喜的进展,筛选出了许多与疾病相关的新型生物标志,不仅为临床疾病的诊断和治疗等提供了新的选择,而且在基础科学、新药研制和疾病预防等方面具有广泛的应用前景[16-18] .本文就SELDI-TOF-MS相关的原理、特点、在临床和基础研究中的应用新进展和未来的发展趋势做一综述.1 ProteinChipâ Array系统和SELDI-TOF-MS的特点1.1 蛋白质芯片系统的组成和原理 蛋白质芯片系统由三部分组成:蛋白质芯片、芯片阅读器和芯片软件.供研究用芯片上有6-10芯池,不同的芯片表面上的化学物质不同,芯片表面分为两大类:一类为化学类表面,包括经典的色谱分析表面,如:结合普通蛋白质的正相表面,用于反相捕获的疏水表面,阴阳离子交换表面和捕获金属结合蛋白的静态金属亲合捕获表面;另一类称为生物类,特定的蛋白质共价结合于预先活化的表面阵列,可以用来研究传统的抗体一抗原反应,DNA和蛋白质作用,受体、配体作用和其他的一些分子之间的相互作用[19] . 根据检测目的不同,可以选用不同的芯片,或者自己设计芯片.将样本和对照点到芯池上以后,经过一段时间的结合反应,用缓冲液或水洗去一些不结合的非特异分子,再加上能量吸收分子(energy absorbing molelule,EAM)溶液,使样本固定在芯片表面.当溶液干燥后,一个含有分析物和大量能量吸收分子“晶体”就形成了.能量吸收分子对于电离来说非常重要.经过以上步骤,就可经把芯片放到芯片阅读器中进行质谱分析. 在阅读器的固定激光束下,芯片上、下移动,使样本上每一个特定点都被“读”到.激光束的每一次闪光释放的能量都聚集在该区一个非常小的点上(focused laser beam,聚焦激光束).这样,每个区都含有丰富的,可寻址(addressable)的位置.蛋白质芯片处理软件精确控制激光寻读过程.当样本受到激发,就开始电离和解除吸附.不同质量的带电离子在电场中飞行的时间长短不同,计算检测到的不同时间,就可以得出质量电荷比,把他输入电脑,形成图像[19].Ball et al [20]采用一种称为人工神经网络(artifical neural network,ANN)的算法处理出现的成千上万的峰,鉴定出三个分子量为13 454、13 457和14 278的生物标记分子,使疾病预测率达到97.1 %.1.2 ProteinChipâ Array芯片和SELDI-TOF-MS的特点 新型蛋白芯片与以往的蛋白芯片不同之处:SELDI-TOF-MS,他是在MALDI(matrix-assisted laser desorption/inionation)[21,22]基础上,改进后实行表面增强的飞行质谱.SELDI-TOF-MS优于MALDI-TOF表现为他不会破坏蛋白质,或使样本与可溶的基质共结晶来产生质谱信号.对SELDI-TOF来说,可以直接将血清、尿液、组织抽取物等不需处理直接点样检测[40] 由于一部分非特异结合的分析物被洗去,因而出现的质峰非常一致,有利于后期分析[23,24] . 与二维电泳相比:二维电泳分析蛋白质的分子量在30 KDa以上时电泳图谱较清楚,对在组织抽提物中占很大比例的低丰度的蛋白质不能被检出;其次,二维电泳胶上的蛋白质斑点很大一部分包含一种以上的蛋白质;而且,二维电泳耗时长,工作量大,对象染色转移等技术要求高,不能完全实现自动化.而SELDI-TOF在200 Da-500 KDa区间都可以给出很好的质谱,对一个样本的分析在几十分钟内就可以完成[19],处理的信息量远远大于二维电泳;对于低丰度物质,即使浓度仅attomole(10-18)的分子,只要与表面探针结合,就可以检测到,这也是二维电泳所不具备的[24,25] . 对于微距阵蛋白芯片来说,需要一种不破坏折叠的蛋白质构象的固定技术,再与另外的蛋白质反应,经检测莹光来观察蛋白质之间的作用[26] .而基于SELDI-TOF-MS的ProteinChip分析蛋白质不需溶解、不需染色、廉价、针对性强. 因而蛋白质芯片仪具有以下优势:(1)可直接使用粗样本,如:血清、尿液、细胞抽提物等[27] .(2)使大规模、超微量、高通量、全自动筛选蛋白质成为可能;(3)他不仅可发现一种蛋白质或生物标记分子,而且还可以发现不同的多种方式的组合蛋白质谱,可能与某种疾病有关[28] (4)推动基因组学发展,验证基因组学方面的变化,基于蛋白质特点发现新的基因.可以推测疾病状态下,基因启动何以与正常状态下不同,受到那些因素的影响,从而跟踪基因的变化[2,14,15] . 其存在的问题:对于不同的样本,根据检测的目标采取或者设计几种芯片,理论上可以把所有的相同性质蛋白质捕获,但是实际上仍有少量的分子没与表面探针结合.使用SELDI-TOF-MS,仅能给出蛋白质的分子量,不能给出C端、N端的序列,也没法知道蛋白质的构型,因此需要将蛋白质充分纯化后,用蛋白酶消化芯片上的蛋白质,分析肽段,再用生物信息学方法鉴定蛋白质序列[18,24] .另外,在国内,该芯片费用较高,分析质谱需要大量后续工作支持.
关于举办“第二届功能蛋白、生物活性肽的开发、应用与大健康产业发展高峰论坛”通知各有关单位: 2017年1月5日国家发展改革委与工业和信息化部联合发布《关于促进食品工业健康发展的指导意见》指出“十三五”期间将积极研究开发功能性蛋白、生物活性肽等保健和健康食品,为功能性蛋白、生物活性肽开发应用带来新的高度。 为进一步推动这一产业健康顺利发展,交流功能性蛋白、生物活性肽有关研究的新技术、新成果和应用新领域,搭建“产学研”之间交流与合作的平台,我单位定于2017年 4月 25日-27日在北京市举办“第二届功能蛋白、生物活性肽的开发、应用与大健康产业发展高峰论坛”。邀请国内外知名专家,为参会者答疑解惑,同时进行科技新成果、新产品展示推介。热忱欢迎全国行业界新老朋友莅临本次论坛。 一、组织机构主办单位:全国医药技术市场协会 国家食品行业生产力促进中心 协办单位:中国化工企业管理协会医药化工专业委员会 支持单位:征集中..........二、时间地点:时间:2017年 4月 25日-27日(25日全天报到)地点:北京市(具体地点、报名后通知)三、会议主要内容及拟邀专家:(采取专场会议形式)* 大会报告1.功能性蛋白、生物活性肽“十三五”相关政策解读2.功能性蛋白、生物活性肽应用的未来发展趋势3.蛋白质组学与质谱分析* 健康食品行业专场1.生物活性肽、功能蛋白配方食品领域研发项目的立项、申报2.生物活性肽、功能蛋白新产品工艺与工程研发3.我国蛋白和生物活性肽食品需求及市场发展趋势分4.功能蛋白、生物活性肽与疾病、保健及免疫作用研究5.蛋白基因组学、微生物学、生物化学及分子生物学6.活性肽功能蛋白营养作用及营养支持7.生物活性肽和蛋白质配方食品安全性研究及评价8.生物活性肽的消化吸收及分布 9.功能性研究、功能性成分分离鉴定10.天然蛋白质资源开发和深加工利用11.食源功能肽产业化技术转化与营养保健开发 12.生物活性肽、功能蛋白检测方法及研究进展13.功能蛋白肽食品生产工艺及安全管理与质量控制/标准建设14.新产品加工工艺开发中的选择及过程控制中关键点15.工艺确认和评价及生产工艺开发的关键步骤* 药品行业专场1.《生物产业发展“十三五”规划》中蛋白质和肽药物发展思路2.“生物类似药研发与评价技术指导原则(试行)”解读3.我国蛋白和多肽药物需求及市场发展趋势分析4.肽及蛋白质类药物传输系统研究5.蛋白和小分子药物设计研究 6.肽和蛋白类药物结构稳定性的研究 7.肽和蛋白质药物临床前安全性研究及评价 8.蛋白质药物构象及生物学活性检测技术9.结构改造的化学策略及生物活性功用、肽合成策略及肽药剂型的应用10.肽和蛋白质类药物微粒制剂的研究和应用 11.毛细管电泳技术在蛋白质及多肽药物研发中的应用 12.蛋白质药物的分离纯化与PEG化学修饰技术 13.蛋白质药物生产工艺 14.多肽、蛋白质药物的大规模生产与制备、成套工艺技术用拟邀嘉宾: 谷瑞增 中国食品发酵工业研究院蛋白功能肽产业研发部 主任 刘新旗 国家“千人计划”专家、北京工商大学 博士 何 梅 北京营养源研究所副所长乐国伟 江南大学食品学院殷腊生 时代(中国)生物活性多肽研究所所长 罗永章 清华大学蛋白质药物北京市重点实验室主任 梁 伟 中科院生物物理研究所蛋白质与多肽实验室任副主任 屈 锋 北京理工大学教授 相关专家报告继续预约中,敬请持续关注! 最终专家日程安排将在会前一周发给报名参会者!四、参会对象:1、健康食品及保健特医食品企业从事开发研究、质量管理的人员、注册专员等高级技术和管理人员;2、新药及仿制药企业从事开发研究、质量管理的人员,负责药品注册文件的编写、审评和注册申报的管理人员;联系人: 马超 电话/传真:010-52706606 手 机:13240487419 电子邮箱:1683101345@qq.com
蛋白质组学研究的一般工具与方法随着人类基因组计划取得巨大的成功和许多物种基因组测序的完成,仅仅靠基因组的序列来试图阐明生命现象是远远不够的,因此,研究重心已经开始从揭示生命的所有遗传信息转移到在分子整体水平对功能的研究上,生命科学已实质性地跨入了后基因组时代。 尽管现在已经有多个物种的基因组被测序,但这些基因组中通常有一半以上基因的功能是未知的。目前功能基因组研究中所采用的策略,如微阵列法(microarray)(Wodicka et al., 1997)、基因芯片(gene chips)(Ramsay et al., 1998)、基因表达序列分析(SAGE)(Velculescu et al., 1995)等,都是从细胞中mRNA的角度来考虑的。但事实上,从DNA、mRNA到蛋白质存在三个层次的调控,mRNA自身也存在着贮存、转运和降解等问题,从mRNA角度考虑,实际上仅包括了转录水平调控,并不能全面代表蛋白质表达水平。实验也证明,组织中mRNA丰度与蛋白质丰度的相关性并不好,尤其对于低丰度蛋白质来说,相关性更差。蛋白质复杂的翻译后修饰,蛋白质的亚细胞定位或迁移,蛋白质-蛋白质相互作用则几乎无法从mRNA水平来判断(曾嵘,夏其昌,2002)。新生肽链合成后存在多种加工、修饰过程,蛋白质间也存在类似于mRNA分子内的剪切、拼接,研究证明基本元件“intein”广泛存在于蛋白质中(Perler et al., 1997)。基因与其编码产物蛋白的线性对应关系只存在于新生肽链而不是最终的功能蛋白质中。 蛋白质是生理功能的执行者和生命现象的直接体现者,对蛋白质结构和功能的研究将直接阐明生命在生理或病理条件下的变化机制;蛋白质本身的存在形式和活动规律,如翻译后修饰、蛋白质间相互作用及蛋白质构象等问题,仍依赖于直接对蛋白质的研究来解决。因此要对生命的复杂活动有全面和深入的认识,必然要在整体、动态、网络的水平上对蛋白质进行研究(钱小红,贺福初,2003)。 蛋白质组学研究中常用的技术体系 方法学上,二维凝胶电泳-质谱仍然是目前最流行和可靠的技术平台(Rabilloud et al., 2000)。其一般过程是:细胞或组织样品——样品制备——二维凝胶电泳(2D-PAGE)分离蛋白质——计算机辅助分析2D-PAGE图象——对感兴趣的蛋白质进行酶解——质谱分析——数据库检索——蛋白质鉴定——分析蛋白质在细胞与组织中的表达情况。 2-D PAGE 样品制备 2D-PAGE 的操作流程基本上实现了程序化。但是,样品制备是一个非常关键与复杂的过程。成功的2D-PAGE取决于对样品中蛋白质有效的抽提和它的溶解性。与核酸不同,目前没有一种通用的方法适用于所有的蛋白质,来源不同的蛋白质都受到自身蛋白质制备方法的挑战。 正确的样品制备方法从收集样品开始时就要防止样品的裂解和被蛋白水解酶降解(Rabilloud et al., 2000)。要尽可能溶解更多的蛋白,并且在2D-PAGE过程中保持它的溶解性,阻止蛋白质的人为修饰。在样品制备过程中,各个实验室也通过实验建立了更为可行的方法。目前通过建立分步提取方法可以有效地提取出更多的蛋白质(兰彦等,2001)。另一种对蛋白质采用预分离的方法称为“多间隔电解法(multi-compartment-electrolyser)”,采用这种方法后,分辨率和胶的质量均明显改善(Herbert et al., 2000)。 但是,由于生物样品的多样性和复杂性,目前所采用的样品制备方法具有局限性。其它物质对蛋白质样品制备存在干扰。核酸通过与蛋白质结合,增加样品黏度而干扰等点聚焦(IEF)分离的效果。当然,通过实验探索,采取一些措施可以减轻它的干扰。例如,在样品制备过程中加入非特异性的核酸酶或RNase与DNase的混合物,在等电聚焦时将每个胶条的电流限制在50mA以内通常可以消除其影响。脂类物质的影响可以通过利用有机溶剂的方法将其去除,但是这常常会导致蛋白质的不可逆沉淀。除了蛋白质的降解之外,糖基化是蛋白质的最重要的人工修饰,样品中的尿素在这一过程中起着非常重要的作用。样品中的尿素在降解的过程中会形成能够与蛋白质的氨基反应的氰酸盐,这种结果会导致蛋白质带有更多的正电荷。所以,在2D-PAGE中要用新鲜的尿素溶液,在等电聚焦过程中要控制温度不能太高(Beranova-Giorgianni, 2003)。但是,目前还没有一种简单有效的方法来去除样品中的多糖。 样品分离和分析 样品制备完成后运用IEF和SDS-PAGE电泳对它进行分离,常采用银染和考马斯亮兰染色即可观察到具有许多蛋白质斑点的凝胶图像。等电聚焦电泳与SDS-PAGE的具体操作步骤已经实现了程序化,均有详细操作流程参考,但是由于样品的不同,不同样品的具体条件还需要试验探索。第二相SDS-PAGE运行结束,染色完毕后,利用计算机软件对凝胶图像进行分析,如PD-QUEST软件,LIPS,HERMES,GEMINI等,对凝胶图像上的蛋白质斑点进行匹配,对图像进行数字化处理等分析(贾宇峰等,2001),对感兴趣的蛋白质采用质谱分析。 低丰度蛋白质的检测 低丰度蛋白在蛋白质组学研究中常常是人们非常感兴趣的,因为细胞或组织中的一些生物活性物质,如细胞分泌的一些活性物质,受体等表达量都非常低。按照一般电泳的上样量,这些小分子是根本看不到的,但如果单纯地增加上样量,细胞或组织中的大量表达的蛋白就会将其覆盖,而且上样量过大也会影响电泳结果。所以对这些低丰度的样品可以进行富集,富集的方法可以通过层析,如亲和层析,离子交换层析等方法,还可以通过利用样品等电点性质等方法将pH范围相近的蛋白质富集(Santoni et al., 2000; Beranova-Giorgianni, 2003)。
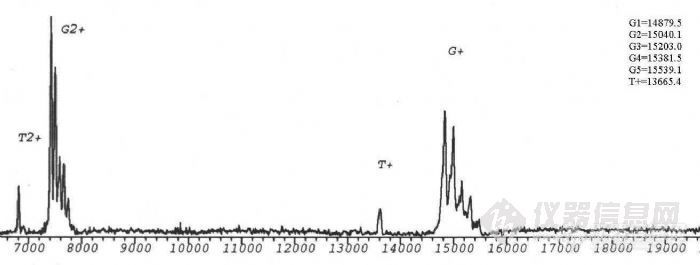
生物质谱在糖蛋白结构分析中的应用项目完成人:桑志红 蔡 耘项目完成单位:国家生物医学分析中心 随着人们对糖蛋白参与生命活动机理的日益深入了解,对天然糖蛋白及重组糖蛋白类药物的分析越来越受到重视。重组糖蛋白类药物的质量控制更是直接关系到药物的疗效及至人类的健康。九十年代以来,随着带有反射功能的基质辅助激光解吸附电离飞行时间质谱(MALDI-TOF-MS)和纳升电喷雾串联质谱(nano-ESI-Q-TOF)等具有软电离方式的现代质谱 技术的发展,质谱以其高灵敏度和强有力的分析混合物的能力,提供了生物大分子的分子量、序列、一级结构信息以及结构转换、修饰等方面的信息,使糖基化分析有了重要的进展。 通常研究糖蛋白的方法是把蛋白链上的寡糖切下来,分别研究蛋白部分和寡糖部分的结构,因此无法研究与两部分共同相关的结构问题,也不能区分不同糖基化位点上切下来的寡糖。自90年代初,国外有人开始用质谱法研究糖蛋白的结构,同时描述了各个位点的不均一性。我们用建立的现代生物质谱技术研究糖蛋白一级结构的方法,将其应用与基因重组糖蛋白的结构分析。为糖蛋白结构分析及基因重组糖蛋白类药物的质量控制提供新的手段。一、 生物质谱研究糖蛋白结构方法的建立实验所用仪器为:1.德国BRUKER 公司的REFLEXIII型基质辅助激光解吸附电离飞行时间质谱仪,N2激光器,波长337nm,线性飞行距离150cm,加速电压2kv。2.英国Micromass 公司Q-TOF型电喷雾串联质谱仪。源温80°C,气体流速40L/h,枪头电压650V,检测频率2.4S,氩气碰撞池压力6*10-5mbar。1. 基质的选择,在MALDI-TOF-MS分析中,基质起着相当重要的作用。不同的基质对不同类的物质响应不同,a-氰基-4-羟基肉桂酸用于测定糖蛋白核糖核酸酶B效果相对较好。2. 糖蛋白分子量的测定,糖蛋白核糖核酸酶B由124个氨基酸组成,在34位Asn处连有一个高甘露糖型N-糖链。由于糖链的微不均一性,与普通蛋白质及核酸不同,其分子离子峰在MALDI-TOF-MS 质谱图上表现为一簇峰,各峰之间约相差一个糖基。正是由于这种微不均一性,使得其分子离子峰变宽,灵敏度降低。糖链分子量越大,峰越宽,灵敏度越低,所以一般只有糖链较短,蛋白的质量不太大的糖蛋白才能测定其平均分子量。用MALDI-TOF可直接测定糖蛋白核糖核酸酶B的平均分子量为 15208.6Da。http://ng1.17img.cn/bbsfiles/images/2011/03/201103211511_284179_1604317_3.jpg3. 糖含量的测定,采用O聚糖酶及内糖苷键酶F分别作用于核糖核酸酶 B,只有内糖苷键酶F能够是其分子量发生变化,表明核糖核酸酶B分子中不存在O-连接糖链存在着N-连接糖链。内糖苷键酶F切断N-糖链五糖核心最内侧的GlcNAc-GlcNAc糖苷键,得到含一个GlcNAc的肽链,减去GlcNAc,可以计算出准确的肽链分子量T=13695.6,与糖蛋白平均分子量之差为糖链的平均分子量G=1513.4,平均糖含量为:(糖链大小/糖蛋白分子量)×100%=9.95%。4. 糖基化位点的确定,研究糖基化类型及糖基化位点的策略:采用蛋白酶酶解与糖苷内切酶酶解相结合的方法,通过酶切前后含糖肽片的位移,结合网上数据库检索,可以确定糖基化类型和糖基化位点。以不同类型的糖苷内切酶作用于糖蛋白(N-糖苷键酶或O-糖苷键酶),在MALDITOF-MS 上观察其质量的变化,可以直接确定糖蛋白中是否含有响应类型的糖链,这是我们确定糖蛋白中糖苷键类型的基础。我们采用先将核糖核酸酶B还原烷基化,加Glu-C酶切,产物再用内糖苷肩酶F酶切,可观察到含糖肽段出现位移,将核糖核酸酶B的肽质量指纹图进行数据库检索,证实发生位移的肽段中含有N-糖链特异连接位点,由此确定34位Asn为糖基化位点。另外我们采用内糖苷键酶F及肽-N-聚糖酶F两种酶进行差位酶切法对含糖肽段进行验证,两种酶酶切后分子离子峰的差值除以GlcNAc的质量,结果就是N-糖基化位点的个数5. 质谱测定氨基酸序列, 我们对核糖核酸酶B肽质量指纹谱中的含糖肽段进行了串联质谱测定,首先在一级质谱图中选择离子4972.23,在串联质谱的碰撞活化室以氩气与其碰撞产生碎片,从碎片的质荷比推算出此肽片中的一段氨基酸序列,检索结果为核糖核酸酶B,从而判断其理论序列是否一致。6. 糖链结构的研究,凝集素对糖肽的亲和提取,进一步分析糖肽序列及糖链结构的关键是含糖肽段的提取。核糖核酸酶B中糖链为高甘露糖型,我们选用对其有特异性吸附的伴刀豆球蛋白对其进行提取利用这种简捷的亲和质谱的方法,对糖肽段进行了分析。建立了亲和质谱分析糖肽类物质的方法,为今后糖肽序列分析及糖链结构分析奠定了基础。二、基因重组糖蛋白人促红细胞生成素(rhEPO)的结构分析。 利用以上建立的方法,我们对样品重组人促红细胞生成素进行了分析,断定此样品为非完全糖基化,样品中只存在N-连接的糖链,无O-糖链。应用酶切法用肽-N-聚糖酶处理后,得到两个含糖肽段,进行数据库检索,测得38位及83位为N-糖基化位点,与文献报道相符,结果可靠。因此,该项课
求达人指点 GB/T24279-2009禁用阻燃剂的标准给出定性选择离子是这样的 例如:磷酸三(β-氯乙基)酯 定量249 定性63:205:143 峰度比100:94:52:50 (CAS号为115-96-8) 买来cas号相同的标准品 按GB/T24279-2009用特征离子扫描(sim)的方法走出来的峰度比为100:10:56:62 其质量数63的峰度差了好多,看到的离子图也不是这种物质。然后用scan走,显示的化合物也不是磷酸三(β-氯乙基)酯 请问这两个问题应该怎么解决
质谱与蛋白质组学蛋白质组学对一个细胞或组织所表达的蛋白质进行的系统分析,而质谱是它的关键性分析工具。在过去的两年中,标准蛋白质组技术中的进展增进了更高水平自动化和敏感性的蛋白质识别技术。另外,新的技术促成了鉴定蛋白质功能相关特性的里程碑性的进展,包括它们的定量和在蛋白质复合物中复杂情况。缩写2DE two-dimensional gel electrophoresis双向凝胶电泳CID collision-induced dissociation碰撞诱导的解离ESI electrospray ionization电喷雾离子化FT-ICR Fourier-transform ion cyclotron resonance傅里叶-变换离子回旋加速器共振ICAT isotope-coded affinity tagsIEF isoelectric focusing等电聚焦MALDI matrix-assisted laser desorption ionization基质辅助的激光解析离子化Q-TOF quadrupole-TOFRP reversed phase反向TOF time-of-flight飞行时间简介蛋白质组学的核心组成是系统识别一个细胞或组织中表达的每一个蛋白质,以及确定每个蛋白质的突出特征(比如,丰度、修饰状态以及在多蛋白质复合体中的复杂状态)。这些分析的技术包括分离蛋白质和肽的分离科学、识别和定量分析物的分析科学和数据管理和分析的生物信息学。它的初步工具包括使用IEF(等电点聚焦)/SDS-PAGE凝胶的高分辨率的双向凝胶电泳(2DE),结合质谱和数据库搜索来分离、识别和定量在一个复合样本中存在的个体蛋白质,最终识别被分离的蛋白质。一个常用的方法用在Fig1中用图解说明。此技术以及由此而来的变化(综述见[1])已经被用来识别和分类在复杂样本中存在的大量蛋白质,并在蛋白质组数据库中呈现它们,该过程我们这里称之为"描述蛋白质组学"比如,Shevchenko等[2]从2D凝胶上系统地鉴定了150个蛋白质。数目庞大的这样的数据库现在可以找到。同样的技术现在已经被作为普遍的发现工具来动态检测一个细胞或组织对外来或内部干扰反应而在蛋白质组中的改变。因为检测动态改变需要精确定量每个被检测成分,我们使用"定量蛋白质组学"来定义。在此报告中,我们总结了自1999年1月至2000年4月来报道的与蛋白质组学和质谱相关的最重要的进展。在核心质谱技术中的进展已经导致2DE为基础的蛋白质组学技术的进一步改进。它们同时又促进了传统凝胶为基础的方法的替代方法,诸如引入以同位素稀释理论为基础的精确蛋白质定量技术和蛋白质复合物的系统分析。蛋白质组分析的MS技术进展在此部分,我们总结了在MS设备、它们的控制和操作中的进展,以及比较质谱数据和序列数据库识别蛋白质所用的搜索工具的进展。随着新型质谱仪的引入,蛋白质组学研究现存类型的质谱仪性能已经显著改进了。在此综述期间最普遍使用的仪器是可以分为两类:单一阶段的质谱仪和串联质谱为基础的系统。单一阶段的质谱仪,最显著的是基质辅助的激光解吸电离(MALDI)飞行时间(TOF)仪器,被用于无数通过肽质谱图谱技术大规模蛋白质识别的项目中。此方法在鉴别表达自小一些的和完全测序的基因组的蛋白质特别成功[3,4]。串联质谱仪器诸如triple quadrpole、离子捕获(ion-trap)和近来引进的混合quadrupole飞行时间(Q-TOF)被常规应用于[url=https://insevent.instrument.com.cn/t/Yp][color=#3333ff]LC-MS[/color][/url]/MS或用电喷雾电离(ESI)来生成肽片段离子谱,以便通过搜寻序列数据库进行蛋白质鉴定。使用仪器控制程序来自动选择肽离子进行碰撞诱导的解离(CID)(数据依赖CID)的不断增多是这些MS/MS仪器的一个明显的趋势。一些新的构造的具有高潜能的质谱仪被引入到蛋白质组学研究中产生深刻影响。两个研究组近来一个MALDI离子源和一个混合Q-TOF耦联了起来[5,6]。Q-TOF提供的质量准确性和敏感性提升了数据库搜寻结果并同时使它成为MS/MS从头测序的当然仪器选择。MALDI Q-TOF构造提供了激动人心的机会进行自动化和高通量应用以及在一个样品盘上存档样品进行日后研究的可能。Medzihradszky等[7]描述了一个不同的混合仪器称之为MALDI TOF TOF。此设备享有许多MALDI Q-TOF的优点,另外能够进行高能量CID和非常快速的扫描速率。傅里叶-变换离子回旋加速器共振(FT-ICR)质谱对于蛋白质组学来说相对陌生。这些设备具有非常高的敏感性和分辨率,质量精确性可以达到1ppm。这些特征被用来在一次分析中测量和定量几百种蛋白质的完整的分子质量[8]。Goodlett等[9]表明FT-MS测量的一个肽的准确质量以及可以容易获得的限制因素能够通过序列数据库搜索被用来识别蛋白质。蛋白质组学如果没有软件工具来进行质谱数据和序列数据库的关联将变得几无可能。现存的数据库搜索程序已经变得越来越成熟和可以(从网络)可获得。另外,引入了新的算法。主要相关程序是Sequest[10],MASCOT[11],PeptedeSearch[12],PROWL[13]和Protein Prospector[14]。在它们中间,Sequest使用CID谱设置了蛋白质识别的实验室标准(benchmark),因为它与边界MS/MS数据工作得最好,并高度可信,可以从整个[url=https://insevent.instrument.com.cn/t/Yp][color=#3333ff]LC-MS[/color][/url]/MS实验中自动分析数据,并不需要任何使用者的破译工作。在所提的程序中,然而,只有Sequest不能在网络上搜索。MASCOT是一个新的、快速、网络可进入和多功能的程序,具有进行肽指纹分析、用部分破译或未破译的CID谱进行数据库搜索的功能。
蛋白质组学研究--生物质谱技术 对分离的蛋白质进行鉴定是蛋白质组研究的重要内容,蛋白质微量测序、氨基酸组成分析等传统的蛋白质鉴定技术不能满足高通量和高效率的要求,生物质谱技术是蛋白质组学的另一支撑技术。 生物质谱技术在离子化方法上主要有两种软电离技术,即基质辅助激光解吸电离(matrix—assisted laser desorption/ionization,MALDl)和电喷雾电离(electrospray ionization,ESl)。MALDI是在激光脉冲的激发下,使样品从基质晶体中挥发并离子化。ESI使分析物从溶液相中电离,适合与液相分离手段(如液相色谱和毛细管电泳)联用。MALDI适于分析简单的肽混合物,而液相色谱与ESI—MS的联用(LC—MS)适合复杂样品的分析。 软电离技术的出现拓展了质谱的应用空间,而质量分析器的改善也推动了质谱仪技术的发展。生物质谱的质量分析器主要有4种:离子阱(iontrap,IT)、飞行时间(TOF)、四极杆(quadrupole)和傅立叶变换离子回旋共振(Fourier transform ion cyclotron resonance,FTICR)。它们的结构和性能各不相同,每一种都有自己的长处与不足。它们可以单独使用,也可以互相组合形成功能更强大的仪器。 离子阱质谱灵敏度较高,性能稳定,具备多级质谱能力,因此被广泛应用于蛋白质组学研究,不足之处是质量精度较低。与离子阱相似,傅立叶变换离子回旋共振(FTICR)质谱也是一种可以“捕获”离子的仪器,但是其腔体内部为高真空和高磁场环境,具有高灵敏度、宽动态范围、高分辨率和质量精度(质量准确度可很容易地小于1mg/L),这使得它可以在一次分析中对数百个完整蛋白质分子进行质量测定和定量。FTICR—MS的一个重要功能是多元串级质谱,与通常的只能选一个母离子的串级质谱方式不同,FTICR—MS可以同时选择几个母离子进行解离,这无疑可以大大增加蛋白质鉴定工作的通量。但是它的缺点也很明显,操作复杂、肽段断裂效率低、价格昂贵等,这些缺点限制了它在蛋白质组学中的广泛应用。MALDI通常与TOF质量分析器联用分析肽段的精确质量,而ESI常与离子阱或三级四极杆质谱联用,通过碰撞诱导解离(collision—induceddissociation,CID)获取肽段的碎片信息。 1. MALDI—TOF—MS (1)MALDI—TOF—MS的技术特点:①具有分离、鉴定双重功能,可用于混合物的分析;②测量范围宽,相对分子质量可达300 000;③精度高,蛋白质相对分子质量测定精度可达0.01%;④灵敏度高,所需样品量少,可达fmol级;⑤分析时间短,5~10rain可完成一次分析;⑥样品制备简便,操作易自动化;⑦对样品要求低,能忍耐较高浓度的盐、缓冲剂和非挥发性杂质,所以特别适合于鉴定二维凝胶电泳分离的蛋白质。 (2)MALDI—TOF—MS的技术改进:近年来MALDI—TOF—MS又有许多新的技术改进,以提高其检测灵敏性和准确度,增强其蛋白质鉴定的功能。如将MALDI离子源与四极杆—飞行时间—串联质谱对接,实现了同一样品在同一质谱仪上的肽指纹图谱与肽序列标签分析同时进行,提高于蛋白质鉴定的速度和准确性。还有MALDI—TOF—TOF—MS等。但这些技术共同的缺点是仪器非常昂贵。 (3)MALDI—TOF—MS的发展方向:①寻求新的基质(如混合基质、室温离子化液体等)和新的制样方法(如超微量进样,n1级);②将新技术应用在分析中(如酶切技术、毫微升溶剂提取技术等);③对质谱仪进行改进或与其他分析仪器联用,如MALDI与傅立叶转换离子回旋共振质谱(MALDI—FTMS)联用能得到更多的蛋白结构信息;④小型化、智能化、简易化及自动化已成为趋势。 2.ESI (1)ESI的优势:①检测范围宽,生物大分子经电喷雾后质荷比大大下降,因而可测相对分子质量高达十几万甚至更高的生物样品;②分辨率和灵敏度高,可达10-15—10-12mol;③不需要特定基质,避免子基质峰的干扰;④适用于结构分析,可分析生物大分子的构象及非共价相互作用,与串联质谱结合可分析蛋白质、多肽的一级结构和共价修饰位点等;⑤自动化程度高,可与液相色谱、毛细管电泳等高效分离手段在线联用;⑥MALDI是脉冲式离子化技术,而ESI是连续离子化技术,检测所需时间更短;⑦ESI常和四极杆质谱联用,仪器价格相对便宜。 (2)ESI的局限性:①对样品中的盐类耐受性差;②对混合物图谱的解析较复杂;③受溶剂的影响和限制很大。目前ESI主要用于亲水生物大分子的分析,较少用于疏水生物样品的分析。总体来说,MALDI和ESI各有长处,有各自的适用范围,是两种互补的技术。 (3)ESI的技术发展:近年来,ESI也有许多新的技术发展。液相色谱与电喷雾质谱连用(LC—ESI—
[摘要] 质谱对于现代蛋白质化学研究是一项重要的技术。也可用于蛋白组分析,在同一时间监控上千种蛋白的表达情况。首先蛋白质混合物可以用双向凝胶电泳分开,再用质谱及随后的蛋白质数据库检索对单个蛋白进行鉴定和分析。最近几年,质谱领域取得了令人鼓舞的进展,使得全自动、高通量的蛋白检测成为可能。质谱也可以用来分析蛋白的转录后调节以及研究蛋白质复合体。本文将介绍目前蛋白组研究中质谱方法的应用并对其优缺点进行讨论。关键词: 质谱;二维电泳(2-DE);蛋白组分析MALDI-TOF1 前 言蛋白组学是研究生物特定组织或细胞内表达的所有蛋白质成分,蛋白质组的一门学科。蛋白组分析一般是用双向凝胶电泳(2-DE),质谱(MS)以及数据检索来完成。2-DE要回顾到20世纪70年代初[1],但是用质谱技术对蛋白质进行鉴定和分析是在近十年内才成为可能。其中最重要的一个原因是新的软离子技术即基质辅助激光解析电离(MALDI)[2]和电喷雾电离(ESI)[3,4]技术的发展和应用,以及样本制备技术和质谱仪的发展等。从数据库中获得成指数上升的序列信息也促进了质谱技术的发展。跟转录组学中的全自动DNA微阵列技术相比,蛋白组分析需要更多的手工操作,尤其在2-DE上会出现很多问题[5,6]。尽管存在很多困难,蛋白组学的重要性还是公认的。DNA微阵列技术是建立在对mRNA稳定水平的检测上,然而蛋白组学研究的对象是细胞中最活跃的因子——蛋白质。最近有研究证明,mRNA的表达和蛋白的水平并非具有相关性[7,8]。蛋白质有转录后修饰过程,并会以不同的形式出现。而这些修饰过程是相应的DNA测序技术所不能预测的。质谱技术在蛋白质修饰分析过程中具有重要的作用。2 质谱对蛋白质的检测质谱仪是由离子源,质量分析,离子检测器,以及数据检索等单元组成。首先,被分析的分子在离子源中被离子化,然后在质量分析器里根据不同的质荷比分开,再对分开的离子进行检测。随着MALDI和EIS的出现,质谱技术已经广泛用来分析蛋白质。质量分析器有很多种,最常用的方法是将飞行时间检测器(TOF)与MALDI或三级四级串联质谱仪联用,或者将四级杆-TOF,离子阱等连接到ESI上。蛋白组分析中,可以用两种不同的方法检测电泳分离后的单个蛋白质。最简单的方法叫做肽指纹谱测定(PMF)[9~13]。这个方法是用特殊的酶对2-DE分离后的蛋白质点进行降解,产生的肽从凝胶中分离出来后再用质谱分析和检测肽的分子量。数据库检测能够产生所有蛋白质理论上的PMF,把它们和质谱分析所获得的肽片进行比较就可以得出结果。第二种方法是在质谱仪中把凝胶分离后的肽分解成片段,产生局部的氨基酸序列(序列标签)。那么就可以用分子量或者序列信息进行数据库检索[14,15]。PMF一般用MALDI-TOF来实现,序列标签可以用串联质谱技术MS-MS进行检测[16~18]。用质谱检测蛋白的灵敏性可以精确到飞摩的水平,甚至已有报道其精确度可以达到阿摩水平[19]。3 用MALDI-TOF进行肽指纹谱分析凝胶分离后的蛋白质鉴定以及肽指纹谱分析是目前质谱实验室常规使用的方法。在进行MALDI分析之前需要最优化的凝胶上消化[20~22]和脱盐过程。胰岛素是最常用的凝胶消化酶,因为它在赖氨酸和精氨酸位点能够特异性切割蛋白质,产生分子量在600~2500Da之间的小分子肽,并能够从凝胶上有效的分离出来。MALDI-TOF是质谱技术中相对简单并很容易使用的一种方式,对样本中的盐和其他污染物有很高的耐受性,这点优于ESI。提取的肽片混合物中较小的片段可以直接沉积在靶板上做PMF分析,剩下的样本可以储存起来作为以后的ESI-MS分析。如果经凝胶分离后所有的肽全用来做MALDI分析,那么样本经脱盐以后将会取得更好的结果。脱盐的过程可以用商品化的试剂盒ZipTip(Millipore),或者在凝胶电泳仪的末端放置一个小的逆向塑料柱来实现[23,24]。PMF中非常重要的一个因素就是质谱测量分子量的准确度[25]。现代的MALDI-TOF仪都具有延迟取样反射器装置,用它们检测的肽段分子量可以精确到10~30ppm。这样的精确度使得4~5个肽段足以清楚的鉴定蛋白质的分子量。MALDI产生的大多数都是单电荷离子,所以它的图谱很容易解释。4 肽质指纹谱分析的自动化为了实现高通量蛋白检测,样本准备,质谱检测,资料分析和数据检索必须实现自动化操作。目前有些实验室用机器人对蛋白质进行自动化取点,凝胶上消化,样本脱盐以及对MALDI平板进行定点测定等。而且,现代化的MALDI-TOF仪完全能够让MALDI检测自动化。资料分析和数据检索过程同样可以自动化。在进行蛋白组分析时,如果在凝胶上不用单个分离就可以对所有蛋白质进行鉴定将是非常具有诱惑力的。为此,研究人员做了很多努力,由Hochestrasser及其同事建立了一套称作分子探测术的方法(molecular scanner)[26,27]。它是将整个2-DE胶上蛋白质样本同时酶解,并将酶解片段一起原位转移至另一张膜上,再直接用MALDI- TOF 质谱对膜上样本进行整体扫描,为实现蛋白质组学研究的自动化、规模化以及临床应用提供了一个崭新的思路和方法。这种思路的优点是显而易见的,不过按照目前的方法,要普及此种技术主要的难点是,质谱扫描一张图谱所需的时间过长,如用0.4mm的精度扫描一张4cm×4cm的膜需55个小时,这就意味着一张16cm×16cm的常用膜的扫描,至少需要36天不间断的工作和大约40G的磁盘空间存储如此庞大的原始数据。5 用ESI-MS/MS的方法创建序列标签只有当被消化的蛋白存在于已有的蛋白或基因组数据库中,并且能从MALDI分析器中得到四五个肽片的数据才能成功进行。如果在EST数据库中只有目的蛋白的部分序列,那么就需要知道肽片的序列信息。创建序列标签的优势在于,用肽的分子量和序列作为数据库检索对象比只用分子量作为检索对象具有更高的特异性。有了序列标签信息后,也可以用EST数据库进行检索[28]。通常来自于一个肽上的序列标签就足以鉴定蛋白质。同MALDI相比,纳升-ESI-MS/MS费时费力,而且在做ESI分析之前必须对样本进行脱盐处理。这些问题可以通过ESI-MS与HPLC联用得到解决。做ESI分析对样本浓度很敏感,用现代化的纳升-HPLC方法可以提供很高的局部肽片凝聚物,分离后的样本可以直接用于质谱分析,而不需要再分解。当分析混合物的时候,在质谱之前做LC分离尤其重要,因为它使得数据依赖的实验(DDE)成为可能,在DDE中,所有的离子都进入检测器进行测量,如果从HPLC到质谱之间出现一个峰的话,软件将自动从质谱转换到MS/MS模式并对产生的离子进行扫描。跟纳升-ESI相比,用这个方法可以从一份标本产生更多的MS/MS数据。Yates等已经详细叙述了如何进行[url=https://insevent.instrument.com.cn/t/Yp][color=#3333ff]LC-MS[/color][/url]/MS全自动蛋白检测的策略[29]。
自80年代以来一系列新的软电离技术如快原子轰击电离 、基质辅助激光解吸电离 、电喷雾电离等发现后,生物质谱技术迅速发展,已成为现代科学研究前沿的热点之一。而其中又以基质辅助激光解吸质谱(MALD I2MS)和电喷雾电离质谱(ESI2MS)应用最为广泛。基质辅助激光解吸质谱灵敏度高、可操作性强且对生物样品中的无机盐和缓冲溶液具有较好包容性;电喷雾电离质谱选择性好、分析质量范围宽、样品消耗量小、易于与各种色谱联用。在生物样品的处理中常常需要用到非挥发性的盐,用于为细胞营造无毒的环境,稳定溶剂化的样品及维持酶的活性等。此外,许多用于分离生物分子的分离方法也需要高浓度的盐和缓冲溶液 。但是,样品处理及分离过程中所用的NaCl、十二烷基磺酸钠、盐酸胍、尿素、甘油、二甲基亚砜等都会影响后续质谱高灵敏的分析 。因为这些不挥发的低分子量污染物会导致复杂加合物的形成,增加噪音及造成明显的信号抑制 。此外,在复杂的组织或细胞蛋白质组中,与疾病和信号传导相关的蛋白质往往是属于低丰度的蛋白质,这些重要的蛋白质由于本身存在的量极少而很难得以有效鉴定 。因此,对蛋白质/多肽样品的预富集处理将是MALD I2MS或ESI2MS得到高质量质谱图的前提,也是成功鉴定蛋白质的关键。该文献主要侧重于相关工作的概述。
[color=#444444]为什么我用离子阱质谱,[url=https://insevent.instrument.com.cn/t/Yp][color=#3333ff]液质联用[/color][/url],测定蛋白质分子量时,得到的蛋白质分子簇不是特别明显,想问一下用什么方法可使得到的蛋白质分子簇更明显些?[/color]
[color=#333333]质谱鉴定样品对蛋白的染色方法要求是什么?[/color]
分子量测定: Q:为什么测定的结果跟理论的结果不一致? A:因为机器测定会有一定的误差,如100000Da的蛋白质最大误差是100Da,如果测定的结果与理论值成倍数关系的可能原因是:0.3倍可能是三电荷峰,0.5倍可能是双电荷峰,原因是样品含碱氨基酸较多;2倍可能是二聚体,3倍可能是三聚体,原因可能是样品中含有过多的二硫键。 Q:为什么我的样品非常纯,但在主峰的附近会有一些小峰?同时在主峰一半或两倍的地方有一个小峰? A:这是因为在激光的扫描的过程中可能会使样品分子损失一些离子,如H2O、NH3等,或者出现样品分子与基质的加合峰,正常的情况下MALDI-TOF单电荷峰占主要,所以有时会在主峰一半或两倍的地方有一个小峰,是双电荷峰和二聚体峰; Q:为什么我的样品纯度、浓度都很好,盐浓度也很低,却得不到结果? A:这只能与蛋白质本身的结构有关,基质不能很好的分离样品,所以希望您能尽可能滇供样品其他的理化质,如酸、碱蛋白、糖蛋白等。的确,有时您的样品的所有质、状态都正常,还是不能得到您想要的结果,这种问题其实已经超出了我们能力的范围,我们仅仅是您整个实验的一个环节,能够做到的是向您提供一个准确的结果。 肽指纹图谱及数据库检索 Q:为什么考马斯染色的结果比银染的结果好。 A:主要是银染的灵敏度较高,得到的蛋白质量较少,再加上在处理样品的过程中银染要脱银,增加洗脱的次数,样品的损失较大,所以噪音和角蛋白污染对目的蛋白的影响较大,对最终的数据库检索也会产生影响 Q:我的PMF的峰值较少,为什么?这些片段数量及峰值是否达到最基本的数据库检索的要求?影响检索的结果吗? A:因为我们用的是胰酶对蛋白质进行降解的,胰酶是一种专一较强的酶,只水解精氨酸、赖氨酸的C端,可能是你的样品的酶解的位点偏多或偏少,偏多酶解为小片段,峰值跟基质在一起难以分辨,偏少肽段的分子量偏大,胶内的样品滞留在胶内,在质谱图上体现不出来,溶液样品可以收集到,但是分子量越大检索的结果越不准确;理论上图谱的峰越多越好,要是都能匹配上那么蛋白的确定程度就更高,相反就越低;一般至少要拿到4个肽段。 Q:如何理解这些片段峰值大小意义,越高(纵坐标值大)越精确吗?以及这些酶解片段的数量及峰值一般应达到什么标准才算比较好?? A:片段峰横坐标的大小的意义是酶解下来的肽段的质量大小,纵坐标表示的收集的该肽段的相对的信号的强度,左边是相对的,右边是绝对的,绝对的越高,抗干扰的能力越好,质谱的精确度由仪器来决定的。我们的仪器的精确度在10ppm以内。 Q:图谱上肽段多于8条,为什么选这8条? A:至于提交检索的肽段的数量是根据肽段的信号强度来选取的,同时在软件处理的过程中会过滤一部分峰, 如胰酶的自解峰等; Q:数据库检索报告中m/zsubmitted,MH+matched两项是什么意思? A:m/z submitted 的意思是您提交给数据库的m/z值(因为质谱仪用m/z来分离样品的), MH+matched指的是与你提交数值相匹配数值,因为在样品在离子化过程中从基质中得到一个质子,所以它会加一个H Q:这两项的分子量和你们的图谱上相应分子量不一致。 A:因为在用图谱数据在数据库检索之前需要通过一个数据的校正和处理,所以图谱上的值与提交检索的值有区别图谱中的峰值是经过一个软件处理后的结果,主要是经同位素峰的合并图谱上的结果会不一至的,例如在你的图上读到的是1519.402,在看图的时候把它给放大处理(只有我这里的图谱处理的软件才能看到),就会发现存在1518.402、1520.402、1521.402、1522.402等相差1的质谱峰,这是由于在天然界中好多物质都存在同位素如O16、O17、O18等,所以在提交给数据库之前会用一个类似于碳水化合物的通式进行校正,所以图谱上读到的数可能会与提交的相差1,还在处理的过程的设置也会影响给出的峰值,但应该在0.1Da以内的;同时在还会进行胰酶自解峰的扣除和空白对照峰的扣除。
亚铁氰化钾和乙酸锌去蛋白,去蛋白后过滤进质谱可以吗?
PS1利用基质辅助激光解吸电离-飞行时间(MALDI-TOF)技术来表征生物分子。样品溶于固定的底物中形成晶体,用激光脉冲使其离子化,离子被加速后通过飞行管时分离,所有离子均可被检测。系统包括三个组成部件:样品点样制备工作站(SymBiot 1)、生物质谱工作站(Voyager-DE PRO)和自动化分析软件(AutoMS-Fit)。SymBiot1 是一个自动样品处理系统,支持亚微升级微量点样,具有快速省时、重现性好的特点;Voyager-DE PRO是为蛋白质组研究专门设计的自动飞行时间质谱分析系统,配有AB公司之专利—延迟检测技术,具有高分辨率、质荷比宽等特点;AutoMS软件可以批处理方式或实时动态方式检索Protein Prospector蛋白数据库或您指定的蛋白数据库,查询参数可以任意设定,检索结果以Microsoft Access格式分类编号及储存。 PS 1技术平台建立伊始便受到了许多蛋白质课题研究组的关注。中国科学院上海生物化学研究所戚正武院士课题组从猪肝中提取某一活性蛋白组分,该组分理化性质不清楚,天然含量十分低,并无相关文献报道。用HPLC分离以后对活性组分的成分不能确定。上海基康生物技术有限公司运用PS 1系统对HPLC分离后的活性组分作了质谱分析,仅在一个工作日内就精确确定该组分由分子量极为相近的几种蛋白质构成,分子量精确度达到10 ppm。后经HPLC再次细分(洗脱梯度增加了2.5倍),证实了质谱的结论。此活性组分曾滤过1kD分子筛,基康的质谱数据纠正了研究人员过去对该活性组分分子量的误判,为研究人员明确实验方向、优化实验步骤提供了强有力的依据。 PS1除了可以进行生物大分子的精确分子量测定,还可用于蛋白的肽指纹图谱分析(peptide mass fingerprint,PMF),提供相关生物信息学服务,并且还可以利用源后衰变(Post Source Decay,PSD)技术来获得样品的MS/MS数据,以得到一级结构信息。PSD方法通常增加了激发激光的功率,使其超过产生一般肽指纹谱图所需功率的阈值,过剩的能量使前体离子在源内离子化之后发生裂解,产生一系列碎片离子,在反射器的作用下,最终可以得到一张连续的碎片离子图谱。经特定的软件分析后,即可在数据库中检索到肽段的氨基酸序列。利用PSD分析技术,还可以对磷酸化,糖基化等翻译后修饰进行定位分析,同样也可以鉴定产生翻译后修饰肽段的蛋白质。Neville et al.(1997)将这一方法成功的用于磷酸肽的序列分析。作为重要的蛋白质鉴定手段之一,PS1的精确度可以达到10 ppm,灵敏度为fmol,分子量检测范围可达到500 kDa,每天可自动分析40-100个样品,适用于大规模“蛋白质组学”研究。
现我详细讲述下我这段时间用该仪器出现的故障,如果哪位前辈和老师能帮我解惑,请不吝赐教,感谢。Thermo DSQⅡ在检测样品的过程中,突然分流流速为0.达不到设定值。出现这种情况,第一时间想到了分流管路堵了,因此把Trace GC分流管用甲醇冲洗之后,出现分流流速控制不灵活了,也就是说有时候能达到设定值,有时候又不能,毫无规律性。同时又增加了新的问题:MS的背景峰出现84的高峰(达到1.0e7)干扰,FC43的峰都没有它高。堵住MS端,质谱里面没有此峰,怀疑GC或载气带进去的,柱子都换了好几个。各位同仁有没有好的简单方法判断哪里出现问题??请赐教《谢谢!》http://ng1.17img.cn/bbsfiles/images/2014/03/201403051206_491914_1616855_3.jpghttp://ng1.17img.cn/bbsfiles/images/2014/03/201403051938_491980_1616855_3.jpg
蛋白质是生物体中含量最高,功能最重要的生物大分子,存在于所有生物细胞,约占细胞干质量的50%以上, 作为生命的物质基础之一,蛋白质在催化生命体内各种反应进行、调节代谢、抵御外来物质入侵及控制遗传信息等方面都起着至关重要的作用,因此蛋白质也是生命科学中极为重要的研究对象。关于蛋白质的分析研究,一直是化学家及生物学家极为关注的问题,其研究的内容主要包括分子量测定,氨基酸鉴定,蛋白质序列分析及立体化学分析等。随着生命科学的发展,仪器分析手段的更新,尤其是质谱分析技术的不断成熟,使这一领域的研究发展迅速。 自约翰.芬恩(JohnB.Fenn)和田中耕一(Koichi.Tanaka)发明了对生物大分子进行确认和结构分析的方法及发明了对生物大分子的质谱分析法以来,随着生命科学及生物技术的迅速发展,生物质谱目前已成为有机质谱中最活跃、最富生命力的前沿研究领域之一[1]。它的发展强有力地推动了人类基因组计划及其后基因组计划的提前完成和有力实施。质谱法已成为研究生物大分子特别是蛋白质研究的主要支撑技术之一,在对蛋白质结构分析的研究中占据了重要地位[2]。 1.质谱分析的特点 质谱分析用于蛋白质等生物活性分子的研究具有如下优点:很高的灵敏度能为亚微克级试样提供信息,能最有效地与色谱联用,适用于复杂体系中痕量物质的鉴定或结构测定,同时具有准确性、易操作性、快速性及很好的普适性。 2.质谱分析的方法 近年来涌现出较成功地用于生物大分子质谱分析的软电离技术主要有下列几种:1)电喷雾电离质谱;2)基质辅助激光解吸电离质谱;3)快原子轰击质谱;4)离子喷雾电离质谱;5)大气压电离质谱。在这些软电离技术中,以前面三种近年来研究得最多,应用得也最广泛[3]。 3.蛋白质的质谱分析 蛋自质是一条或多条肽链以特殊方式组合的生物大分子,复杂结构主要包括以肽链为基础的肽链线型序列[称为一级结构]及由肽链卷曲折叠而形成三维[称为二级,三级或四级]结构。目前质谱主要测定蛋自质一级结构包括分子量、肽链氨基酸排序及多肽或二硫键数目和位置。 3.1蛋白质的质谱分析原理 以往质谱(MS)仅用于小分子挥发物质的分析,由于新的离子化技术的出现,如介质辅助的激光解析/离子化、电喷雾离子化,各种新的质谱技术开始用于生物大分子的分析。其原理是:通过电离源将蛋白质分子转化为[url=https://insevent.instrument.com.cn/t/Mp]气相[/url]离子,然后利用质谱分析仪的电场、磁场将具有特定质量与电荷比值(M/Z值)的蛋白质离子分离开来,经过离子检测器收集分离的离子,确定离子的M/Z值,分析鉴定未知蛋白质。 3.2蛋白质和肽的序列分析 现代研究结果发现越来越多的小肽同蛋白质一样具有生物功能,建立具有特殊、高效的生物功能肽的肽库是现在的研究热点之一。因此需要高效率、高灵敏度的肽和蛋白质序列测定方法支持这些研究的进行。现有的肽和蛋白质测序方法包括N末端序列测定的化学方法Edman法、C末端酶解方法、C末端化学降解法等,这些方法都存在一些缺陷。例如作为肽和蛋白质序列测定标准方法的N末端氨基酸苯异硫氰酸酯(phenylisothiocyanate)PITC分析法(即Edman法,又称PTH法),测序速度较慢(50个氨基酸残基/天);样品用量较大(nmol级或几十pmol级);对样品纯度要求很高;对于修饰氨基酸残基往往会错误识别,而对N末端保护的肽链则无法测序[4]。C末端化学降解测序法则由于无法找到PITC这样理想的化学探针,其发展仍面临着很大的困难。在这种背景下,质谱由于很高的灵敏度、准确性、易操作性、快速性及很好的普适性而倍受科学家的广泛注意。在质谱测序中,灵敏度及准确性随分子量增大有明显降低,所以肽的序列分析比蛋白容易许多,许多研究也都是以肽作为分析对象进行的。近年来随着电喷雾电离质谱(electrospray ionisation,ESI)及基质辅助激光解吸质谱(matrix assisted laser desorption/ionization,MALDI)等质谱软电离技术的发展与完善,极性肽分子的分析成为可能,检测限下降到fmol级别,可测定分子量范围则高达100000Da,目前基质辅助的激光解吸电离飞行时间质谱法(MALDI TOF MS)已成为测定生物大分子尤其是蛋白质、多肽分子量和一级结构的有效工具,也是当今生命科学领域中重大课题——蛋白质组研究所必不可缺的关键技术之一 [5] 。目前在欧洲分子生物实验室(EMBL)及美国、瑞士等国的一些高校已建立了MALDI TOF MS蛋白质一级结构(序列)谱库,能为解析FAST谱图提供极大的帮助,并为确证分析结果提供可靠的依据[6]。
生物质谱技术帮助发现精神分裂症特征蛋白 (2006-12-28 9:34:47,新闻来源:人民网) 日前,四川泸州医学院附属医院硕士研究生姜伟,在导师王开正教授指导下,利用表面增强激光解析电离飞行时间质谱技术,发现了一组对精神分裂症的早期预警、鉴别诊断、治疗观察具有重要意义的蛋白标志物。 目前鉴别诊断精神分裂症缺乏有效的病理学、影像学和实验诊断依据,也缺乏客观的检查指标用于早期诊断,这也是造成对某些精神分裂症的评定产生分歧、鉴定困难的原因之一。 姜伟等人应用获得诺贝尔化学奖、高灵敏度高解析度的表面增强激光解析电离飞行时间质谱技术,检测精神分裂症血清蛋白质指纹图谱,将40名精神分裂症患者血清与44名其他人对照血清随机分为训练集和验证集,将筛选训练集精神分裂症差异蛋白建立人工神经网络诊断模型,再将验证集验证该模型的诊断效率。结果发现精神分裂症患者与对照组血清蛋白质指纹图谱有15个差异表达的蛋白质荷比峰,筛选出其中6个有明显表达差异的标志蛋白,建立的人工神经网络诊断模型对精神分裂症的诊断灵敏度和特异性分别为95.0%和95.8%,阴性预测值和阳性预测值分别为95.8%和95.0%。姜伟等人依据这一结果认为,精神分裂症患者血清具有高表达的特征蛋白,建立的人工神经网络模型为精神分裂症的实验诊断提供了一种蛋白组学的新方法。这种方法检查过程快速、简便,不会破坏所测定的蛋白质,结果可靠,可重复检测。 ————2006.12.27健康报也有报道--------------------------------------------------------------------另:两毫升血5小时鉴别精神分裂症 作者:周丽 文章来源:泸州晚报 点击数:0 更新时间:2006-12-20 只需要从体内抽取两毫升鲜血,经过5个小时的实验室检测,就能诊断是否患有精神分裂症。昨(19)日记者获悉,如此简便的方法已经在我市投入使用,这也是西南地区首次将诺贝尔化学奖应用到精神分裂症的实验室诊断中。 据悉,该技术在泸医附院检验科正式投入使用。泸医附院检验科主任王元正告诉记者,泸医附院引进的表面增强激光解析电离飞行时间质谱技术是获得2002年诺贝尔化学奖的高新技术,通过对患者的抽血检测,发现了一组对精神分裂症的早期预警、鉴别诊断、治疗观察具有重要意义的蛋白标志物。 而泸医附院引进的这种技术,将精神分裂症患者血清蛋白指纹图与健康人做差异蛋白组学分析,利用计算机程序找到了精神分裂症患者特征表达的蛋白质,将6个蛋白的相对含量建立人工神经网络诊断模型并进行盲法验证。结果显示,对精神分裂症的诊断灵敏度为95.0%,特异性为95.8%。整个实验检测过程只需要5个小时。 目前,全国对此技术的利用并不广泛,泸医附院在西南地区率先引进使用。这种技术还用于肿瘤、心血管疾病、风湿免疫性疾病、感染性疾病等疑难疾病的鉴别诊断,特别是对各种肿瘤的早期诊断灵敏度和特异性都在95%以上。
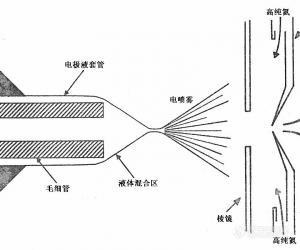
电喷雾电离质谱及其在蛋白质化学研究中的应用 桑志红综述 杨松成审校 (国家生物医学分析中心 北京 100850) 摘要 本文综述了电喷雾电离质谱及其在蛋白质化学研究中的应用。由于电喷雾电离质谱可产生多电荷峰,因此大大扩大了检测的分子质量范围,同时灵敏度高,另外它可与HPLC及高效毛细管电泳分离技术联用,扩大了质谱在蛋白质化学研究中的应用。关键词 电喷雾电离;质谱;蛋白质化学 在有机化合物结构的鉴定中,质谱、核磁、红外及紫外等分析手段,从不同的侧面提供了化合物的结构信息。质谱以质量分析为基础,灵敏度高,可提供化合物的分子量、分子式(高分辨质谱)以及一些有关的结构信息。经典的有机质谱要求待测物能气化,有一定纯度,热稳定性好等条件,因此,极性高,不易气化,热不稳定以及不纯的化合物难以用经典质谱测定。近年来随着有机质谱在质谱硬件、软件、电离技术的发展,以及与各种分离方法相联(如色质联用技术)的接口的不断完善,扩大了化合物的检测范围,在分子量测定方面,已从化学小分子扩展到生物大分子,可测定的分子量达到几十万道尔顿。质谱有多种电离方法,包括场解吸、等离子体解吸、激光解吸、快速粒子轰击、热喷雾电离和大气压电离等。每一种电离方法都有一定的分子量检测范围,一般认为热喷雾的分子量检测最大范围约8ku,快原子轰击为25ku。但是随着分子质量的增加,所有分析方法的灵敏度均有所下降。电喷雾电离质谱(ESI-MS)由于可以产生多电荷峰,与传统的质谱相比扩大了检测的分子质量范围,同时提高了灵敏度,使一种M/Z限制在一定范围的四极质谱,就可以分析分子质量超过200ku的蛋白质[1]。另外ESI-MS方法产生一系列的多电荷峰,可以得到准确的分子量,它还可与HPLC和高效毛细管电泳(CE)分离方法相连接,扩大了质谱在生物领域的应用。电喷雾现象的出现可以追溯到两个世纪之前,但真正把电喷雾作为一种电离方法的创新性的研究是由Dole等在大约30前开始的,他们研究的目的是用电喷雾来产生气态大离子。1984年Yamashita等把大气压电喷雾电离技术与四极质谱结合起来,同年,Alexandror把它和磁质谱结合起来。1988年Fenn研究小组报道了用ESI-MS得到了带有45个正电荷分子量为40ku的蛋白质,随后ESI-MS在生物大分子的研究领域进入了一个全新的发展阶段。到目前为止,该法已经能够分析质量范围大约在200ku的蛋白质。1.电喷雾电离过程[img]http://ng1.17img.cn/bbsfiles/images/2006/08/200608250906_24655_1237095_3.jpg[/img]附图 电喷雾质谱的电离接口示意图 如附图所示,在毛细管管口加一高电压,作用于经喷雾头进入离子化室的溶液,再将3~6kV的电压加到毛细管和相对的电极之间,电压导致毛细管末端的液滴表面的电荷增强,高电压导致液体表面分裂和多电荷液滴的形成,与毛细管子相对的电极携带的正或负电荷以产生正或负电荷液滴。对于电喷雾的整体而言,带电液滴的形成是整个电喷雾过程的第一步,而接下来的离子化是进行电喷雾分析的关键。而带电液滴形成分子离子的机制还不清楚。Iribane和Thomson提出场辅助离子蒸发假设。在这种模型中,处于液滴表面的离子是由于带电液滴的溶剂在空气中蒸发,当场力在液滴表面达到临界点时由液相直接进入[url=https://insevent.instrument.com.cn/t/Mp]气相[/url]完成离子化的过程。而Rollgen等提出了不同的假设机理,他认为当液滴在大气压下蒸发时,随着溶剂的蒸发,由于液滴直径变小,液滴表面电荷密度增加,当液滴表面电荷达到雷利极限(Raleigh limit),液滴进一步裂变,再次达到雷利极限,再一次“爆炸”,如此循环,当溶剂从小液滴中完全蒸发后形成分子离子。Abbas和Latham的实验证实了从液滴生成分子离子的这一假设。 对于大分子化合物离子化过程的形成可用带电残渣模型理论加以解释,该理论认为大分子[url=https://insevent.instrument.com.cn/t/Mp]气相[/url]离子的形成是基于溶剂蒸发,由库仑爆裂辅助及较小液滴的相互排斥而导致液滴形成仅含有一个分子离子的[url=https://insevent.instrument.com.cn/t/Mp]气相[/url]离子,此过程所需能量很低,不会导致分子裂解。但是通过离子源内离子传输区的碰撞诱导解离(CID)电压设置可得到一些有效的碎片离子,但这只对一些不稳定结构有效,并且ESI-MS的CID质谱与电子轰击质谱(EI)及快原子轰击质谱(FAB)有一定的不同,可能是由于前者的开裂环境比EI及FAB源更为复杂,因此在对化合物结构的获得上有一定的制约。随着现代技术的发展,电喷雾与串联质谱(MS-MS)相连,即能为化合物提供很多结构信息。 2.电喷雾电离质谱的分子质量的检测 由于ESI-MS的分子离子状态不是由于裂解(除非在高能量下发生碰撞诱导解离进入真空系统)而得到,因此对于生物大分子的分子量测定变得比较容易。ESI-MS得到的是一簇多电荷的质谱峰群,它的分子质量的测定可从以下假设中得出:(1)两相邻峰相差一个电荷;(2)电荷是由于分子离子质子化形成的。任何两个峰都可有效地测定分子质量。方程式(1),(2)描述了分子质量Mr和多电荷离子(P1,P2)以及它们各自所带电荷(Z1,Z2)之间的关系。P1Z1=Mr+MaZ1=Mr+1.0079Z1 (1) P2Z2=Mr+1.0079Z2 (2)当P2P1时解(1)、(2)方程,得 Z1=J(P2-1.0079)/(P2-P1) (3)通过Z1可得到准确的分子质量。于是可将一簇多电荷峰的质谱图转化成化合物[M+H]+或[M+H]-的质谱图。
一、 前言基因工程已令人难以置信的扩展了我们关于有机体DNA序列的认识。但是仍有许多新识别的基因的功能还不知道,也不知道基因产物是如何相互作用从而产生活的有机体的。功能基因组试图通过大规模实验方法来回答这些问题。但由于仅从DNA序列尚不能回答某基因的表达时间、表达量、蛋白质翻译后加工和修饰的情况、以及它们的亚细胞分布等等,因此在整体水平上研究蛋白质表达及其功能变得日益显得重要。这些在基因组中不能解决的问题可望在蛋白质组研究中找到答案。蛋白质组研究的数据与基因组数据的整合,将会在后基因组研究中发挥重要作用。目前蛋白质组研究采用的主要技术是双向凝胶电泳和质谱方法。双向凝胶电泳的基本原理是蛋白质首先根据其等电点,第一向在pH梯度胶内等电聚焦,然后转90度按他们的分子量大小进行第二向的SDS-PAGE分离。质谱在90年代得到了长足的发展,生物质谱当上了主角,蛋白质组学又为生物质谱提供了一个大舞台。他们中首选的是MALDI-TOF,其分析容量大,单电荷为主的测定分子量高达30万,干扰因素少,适合蛋白质组的大规模分析。其次ESI为主的LC-MS联机适于精细的研究。本文将简介几种常用的生物质谱技术,并着重介绍生物质谱技术在蛋白质组学各领域的应用。此贴与http://bbs.instrument.com.cn/shtml/20081130/1613156/重复,请版友在发帖前先搜索一下,以免重复发帖,谢谢!
一、 前言[1,2]基因工程已令人难以置信的扩展了我们关于有机体DNA序列的认识。但是仍有许多新识别的基因的功能还不知道,也不知道基因产物是如何相互作用从而产生活的有机体的。功能基因组试图通过大规模实验方法来回答这些问题。但由于仅从DNA序列尚不能回答某基因的表达时间、表达量、蛋白质翻译后加工和修饰的情况、以及它们的亚细胞分布等等,因此在整体水平上研究蛋白质表达及其功能变得日益显得重要。这些在基因组中不能解决的问题可望在蛋白质组研究中找到答案。蛋白质组研究的数据与基因组数据的整合,将会在后基因组研究中发挥重要作用。目前蛋白质组研究采用的主要技术是双向凝胶电泳和质谱方法。双向凝胶电泳的基本原理是蛋白质首先根据其等电点,第一向在pH梯度胶内等电聚焦,然后转90度按他们的分子量大小进行第二向的SDS-PAGE分离。质谱在90年代得到了长足的发展,生物质谱当上了主角,蛋白质组学又为生物质谱提供了一个大舞台。他们中首选的是MALDI-TOF,其分析容量大,单电荷为主的测定分子量高达30万,干扰因素少,适合蛋白质组的大规模分析。其次ESI为主的[url=https://insevent.instrument.com.cn/t/Yp][color=#3333ff]LC-MS[/color][/url]联机适于精细的研究。本文将简介几种常用的生物质谱技术,并着重介绍生物质谱技术在蛋白质组学各领域的应用。
[color=#444444]质谱最高峰是加钠离子的峰 谱图怎么解析啊?MS要写(M+Na)吗?还是写(M+1)的那个数值啊?请高手指教。[/color]
20世纪基因组学研究取得的巨大成就为蛋白质组学的发展奠定了基础。蛋白质组学是从整体水平上分析生命体、组织或细胞的蛋白质组成及其活动规律的科学,以基因表达产物为研究对象,延伸了基因组学研究深度,更深层次地揭示了生命活动规律。蛋白质组学的研究内容主要包括蛋白质表达存在方式(修饰形式)的鉴定、结构与功能分析、蛋白质定位、蛋白质差异表达以及蛋白质间相互作用分析等[1]。目前蛋白质组学研究技术主要包括:二维电泳技术、蛋白质芯片技术、质谱技术等[2]。其中,二维电泳技术是早期蛋白质组学的重要技术之一,但是由于实验步骤多,耗时长,重复性差等特点,已经逐步被新型技术所取代。蛋白质芯片技术是将多种蛋白质纯品点于芯片表面,形成蛋白质矩阵进行免疫等标记反应,主要受限于很多蛋白质无法获得纯品而不能用于芯片制备。质谱技术由于灵敏度高、特异性强、分析范围宽等优点逐渐成为蛋白质组学的主要研究手段,可以对特定生命过程中的功能性蛋白质分子进行定性和定量检测,因此在基础科研和临床研究中得到了广泛的应用[3,4]。一、基于质谱的蛋白质组学技术1.基于质谱的蛋白质组学定性技术:蛋白质定性鉴定的基本原理在于:蛋白质组的基本序列已经通过基因组学信息获得,可以用来鉴定多肽的氨基酸序列,并且获得多肽与蛋白质的对应关系[1],即质谱提供的多肽碎片数据可以与蛋白质数据库自动匹配来确定多肽序列与蛋白质归属。基本技术策略分为:(1)自上而下(Top–down)策略[5],即完整蛋白质在质谱中进行分析,可以提供完整蛋白质的质量数,但是由于质谱仪受到质量分析范围的限制,此方法在常规实验室不易实现。(2)自下而上(Bottom–up)策略[6],即蛋白质被蛋白酶水解成多肽,然后对多肽进行质谱分析和碎裂。基于这条策略的大致步骤为:蛋白质样品首先经过酶解降解为多肽,然后对多肽进行色谱–质谱分离与鉴定,最后通过搜索引擎(MASCOT:http://www.matrixscience.com/server.html, SEQUEST:http://fields.scripps.edu/sequest等)在公共蛋白质组学数据库(SWISS–PORT: http://web.expasy.org/groups/swissprot, NCBI:http://www.ncbi.nlm.nih.gov/pubmed等)中自动完成质谱数据的解析,确定多肽序列与蛋白质种类。该技术灵敏度高,特异性好,仪器自动化程度高,可以鉴定出生物样品中成千上万种蛋白质,被认为是大规模、高通量蛋白质定性检测的首选方法。2.基于质谱的蛋白质组学相对定量技术:对于大多数生命科学和医学研究来说,仅完成样品中蛋白质组的定性研究是远远不够的,还需要对蛋白质组进行定量分析。由于组学的研究对象是多个蛋白质,单次检测很难实现所有蛋白质的绝对定量,因此蛋白质组学定量多为相对定量检测。蛋白质组学定量的质谱技术包括谱图计数、质谱峰强度定量、同位素定量技术等。其中使用同位素作为内标定量的方法是目前质谱定量的最佳手段,即对整体蛋白质组进行同位素标记,并使用每一种天然蛋白质与同位素蛋白质的比值进行相对定量分析。主要分为细胞层面标记和蛋白质层面标记两种技术路线:(1)细胞层面标记的细胞培养氨基酸稳定同位素标记(stable isotope labeling with amino acids in cell culture,SILAC)方法[7]:即在两种细胞样品中分别加入轻重同位素标记的培养基,经过传代培养后,两种细胞样品中的全部蛋白质中分别嵌合了轻重同位素,可以在质谱上根据同位素的不同质荷比直接判断样品来源并进行定量比对。(2)蛋白质层面标记:使用含有同位素的小分子与样品全部蛋白质直接标记,如同位素标记相对和绝对定量技术(isobaric tags for relative and absolute quantification,iTRAQ)[8]、同位素编码亲和标记(isotope–coded affinity tag,iCAT) [9]、18O标记[10]等方法,此类方法使用带有稳定同位素的小分子与特定氨基酸侧链反应,使得多个样品可以分别连接含有不同同位素个数(多至8个)的小分子,从而产生一级数据相同但是二级数据不同的质谱谱图,通过二级谱图强度比对进行多个样品的定量分析。3.基于质谱的目标蛋白质绝对定量技术:质谱技术对目标蛋白质的绝对定量检测主要通过质谱多反应监控技术与同位素多肽内标技术联用来实现[11]。该方法首先选定目标蛋白质的一个或多个多肽,合成序列相同但含有稳定同位素的多肽作为内标,定量加入样品中,通过监测特定多肽及其同位素多肽的质谱峰强度进行比对和计算获得目标蛋白质的定量值。质谱多反应监控技术通过进行母离子筛选与子离子筛选等二次选择过程,筛选出目标蛋白质,而非目标蛋白质由于无法通过筛选达到检测器,极大降低了噪音干扰。因此,此方法针对性强,本底噪音低,是目前质谱技术中定量能力最好的一种,可以控制变异系数小于15%,检测限低至纳克每毫升,适合血液、组织等临床样品的定量检测[11]。二、质谱技术发现肿瘤蛋白质标志物质谱技术作为一项强有力的研究工具在科学研究中发挥着巨大的作用,特别在肿瘤相关研究中,目前已经获得美国食品药品监督管理局(Food and Drug Administration,FDA)批准的肿瘤标志物包括多种蛋白质前列腺特异性抗原(prostate–specific antigen, PSA), 癌胚抗原(carcinoembryonic antigen CEA), 人类表皮生长因子受体2(human epidermal growth factor receptor 2,Her–2), 人绒毛膜促性腺激素(human chorionic gonadotropin, HCG), 糖类抗原CA125等,均揭示了蛋白质与肿瘤发生发展密切相关。这些已有成果极大促进了质谱技术在肿瘤蛋白质标志物研究中的应用,并取得了标志性进展。例如:美国约翰霍普金斯大学的Chan课题组发现了新型卵巢癌蛋白质标志物,他们使用表面增强激光解析电离质谱技术(surface enhanced laser desorption and ionization time–of–flight mass spectrometry, SELDI–TOF MS)技术对503个妇女的血清进行了蛋白质组学的分析[12],在随后的大量临床验证中最终确定CA125、β2微球蛋白,转铁蛋白,甲状腺运载蛋白和载脂蛋白A1的联合检测可以作为卵巢癌的新型临床诊断指标。2009年9月该试剂盒OVA1(商品名称:http://ova–1.com)获得了美国FDA的认证,进入临床使用,被认为是国际肿瘤蛋白质标志物研究的重要标志性成果。同时,肿瘤仍然是国际上致死率最高的疾病之一,缺乏早期检测技术和有效治疗方案,临床中还存在着大量问题需要解决,新型标志物的研发迫在眉睫。由于肿瘤蛋白质标志物研究的难度大,风险高,因此近十年来仅有几例试剂盒获得了美国FDA批准,进入临床使用。大量标志物研究还停留在论文研究水平,其中临床问题、研究思路和技术方案的选择直接关系到研究的成功与否。1.临床问题选择:在肿瘤蛋白质标志物研究中,临床问题的选择是研究核心。在肿瘤研究中,需要解决的临床问题往往包括肿瘤早期检测、肿瘤分期检测、治疗方案与药物选择、疗效评估等多个方面。研究者需要根据不同肿瘤的临床情况,具体分析并凝练不同肿瘤的主要临床问题。例如,对于病程发展快、五年存活率低、没有有效手术或化疗手段的肿瘤,早期诊断是研究重点,如胰腺癌、卵巢癌、肺癌等;对于病程发展慢、手术效果明显的肿瘤,肿瘤的愈后与复发是需要关注的问题,如前列腺癌、肠癌等;还有一些肿瘤有特殊的检测需求,如乳腺癌虽然有临床有效的雌激素受体(estrogen receptor,ER),孕激素受体(progesterone receptor,PR),HER2等基因标志物,可以进行药物靶点治疗,但是三阴性乳腺癌的检测还缺乏有效的标志物与治疗方案。因此,在肿瘤蛋白质标志物研究实验开展之前,明确临床问题,并以此确定临床样品入组标准,是研究成功的核心基础。2.研究思路设计:不同于基础科学实验,临床实验需要在大量样本中进行实验结果的验证,因此肿瘤蛋白质标志物研究往往包括新型标志物发现和验证两部分。标志物发现实验是在疾病组和对照组之间进行蛋白质组学分析,鉴定样本中的未知蛋白质组并进行相对定量比较,分析数据选择出在两组样本中差异最大的一个或几个蛋白质作为新型标志物的候选物。随后,标志物验证实验在大量未知样本中进行蛋白质候选物的定量检测,使用发现实验中建立的区分标准进行判读,计算检测灵敏性(sensitivity)和特异性(specificity)。有效的蛋白质标志物研究往往需要发现与验证的两步设计思路来相互保证。3.技术方案选择:根据蛋白质标志物研究的两步设计思路,发现实验中使用基于质谱的蛋白质组学定性技术与相对定量技术对样本中的大量未知蛋白质进行分析,获得标志物候选物名单。验证实验中根据已有名单,进行目标蛋白质(非蛋白质组学)的精确定量检测。这几种质谱技术的配合使用,可以满足不同实验情况和目的,最终实现新型蛋白质标志物的成功研发。三、展望质谱技术是现阶段蛋白质组学研究的核心技术,具有灵敏度高、特异性强、分析通量大等优势,特别是其与同位素内标的联合使用,大大提高了质谱定量能力,因此在多种肿瘤标志物研究中取得了突破性进展并被广泛应用。目前,大量肿瘤蛋白质标志物候选物已经通过使用质谱技术被从血液、组织、体液中筛选出来,预计在完成大规模临床验证后可以作为新型标志物在临床使用,促进肿瘤检测水平的发展。同时值得注意的是,质谱技术还不具备进行蛋白质组的绝对定量能力。相对于免疫等传统蛋白质检测技术,仪器昂贵,操作复杂,自动化程度低,这些因素决定了质谱目前适用于蛋白质的临床研究,但不适用于蛋白质的临床检验,这是质谱技术面临的重要挑战之一。参考文献[1]何华勤. 简明蛋白质组学[M]. 北京:中国林业出版社, 2011:1,76,85-95,119,125-138.[2]RuediA, MatthiasM. Mass spectrometry-based proteomics[J]. Nature, 2003, 422(13):198-207.[3]甄艳, 施季森. 质谱技术在蛋白质组学研究中的应用[J]. 南京林业大学学报:自然科学版, 2011, 35(1):103-108.[4]孙瑞祥, 付岩, 李德泉,等. 基于质谱技术的计算蛋白质组学研究[J]. 中国科学E辑信息科学, 2006, 36(2):222-234.[5]WhiteleggeJ,HalgandF,SoudaP, et al. Top-down mass spectrometry of integral membrane proteins [J]. Expert Review Proteomics, 2006, 3(6):585-596.[6]ChaitBT. Mass spectrometry:bottom-up or top-down? [J]. Science, 2006, 314(5796):65-66.[7]TranDT, AdhikariJ, FitzgeraldMC. StableIsotope Labeling with Amino Acids in Cell Culture (SILAC)-based strategy for proteome-wide thermodynamic analysis of protein-ligand binding interactions [J]. Mol Cell Proteomics, 2014,13(7):1800-1813.[8]DytfeldD, KandarpaM, StrahlerJR, et al. Proteomic Profiling of Multiple Myeloma (MM) Cells Using iTRAQ and Label-Free Quantitative Proteomics for the Prediction of Complete or near Complete Response (CR/nCR) In Frontline Treatment with Lenalidomide, Bortezomib, and Dexamethasone [J]. Blood, 2010, 116(21):271-272.[9]García-SantamarinaS, BoronatS, DomènechA, et al. Monitoring in vivo reversible cysteine oxidation in proteins using ICAT and mass spectrometry [J]. Nat Protoc,2014,9(5):1131-1145.[10]MirzaSP, GreeneAS, OlivierM. 18O labeling over a coffee break:a rapid strategy for quantitative proteomics [J]. J Proteome Res, 2008,7(7):3042-3048.[11]曹冬, 张养军, 钱小红. 基于生物质谱的蛋白质组学绝对定量方法研究进展[J]. 质谱学报, 2008, 29(3):185-190.[12]ZhangZ, BastRC, YuY,et al. Three biomarkers identified from serum proteomic analysis for the detection of early stage ovarian cancer[J]. Cancer Res,2004,64(16), 5882-5890.
传统的蛋白鉴定方法,如免疫印迹法、内肽的化学测序、已知或未知蛋白的comigration分析,或者在一个有机体中有意义的基因的过表达通常耗时、耗力,不适合高流通量的筛选。 目前,所选用的技术包括对于蛋白鉴定的图象分析、微量测序、进一步对肽片段进行鉴定的氨基酸组分分析和与质谱相关的技术。1 图象分析技术(Image analysis)“满天星”式的2-DE图谱分析不能依靠本能的直觉,每一个图象上斑点的上调、下调及出现、消失,都可能在生理和病理状态下产生,必须依靠计算机为基础的数据处理,进行定量分析。 在一系列高质量的2-DE凝胶产生(低背景染色,高度的重复性)的前提下,图象分析包括斑点检测、背景消减、斑点配比和数据库构建。 首先,采集图象通常所用的系统是电荷耦合CCD(charge coupled device)照相机;激光密度仪(laser densitometers)和Phospho或Fluoroimagers,对图象进行数字化。 并成为以象素(pixels)为基础的空间和网格。 其次,在图象灰度水平上过滤和变形,进行图象加工,以进行斑点检测。 利用Laplacian,Gaussian,DOG(difference of Gaussians) opreator使有意义的区域与背景分离,精确限定斑点的强度、面积、周长和方向。图象分析检测的斑点须与肉眼观测的斑点一致。 在这一原则下,多数系统以控制斑点的重心或最高峰来分析,边缘检测的软件可精确描述斑点外观,并进行边缘检测和邻近分析,以增加精确度。 通过阈值分析、边缘检测、销蚀和扩大斑点检测的基本工具还可恢复共迁移的斑点边界。 以PC机为基础的软件Phoretix-2D正挑战古老的Unix为基础的2-D分析软件包。 第三,一旦2-DE图象上的斑点被检测,许多图象需要分析比较、增加、消减或均值化。 由于在2-DE中出现100%的重复性是很困难的,由此凝胶间的蛋白质的配比对于图象分析系统是一个挑战。 IPG技术的出现已使斑点配比变得容易。 因此,较大程度的相似性可通过斑点配比向量算法在长度和平行度观测。 用来配比的著名软件系统包括Quest,Lips,Hermes,Gemini等,计算机方法如相似性、聚类分析、等级分类和主要因素分析已被采用,而神经网络、子波变换和实用分析在未来可被采用。 配比通常由一个人操作,其手工设定大约50个突出的斑点作为“路标”,进行交叉配比。 之后,扩展至整个胶。例如:精确的PI和MW(分子量)的估计通过参考图上20个或更多的已知蛋白所组成的标准曲线来计算未知蛋白的PI和MW。 在凝胶图象分析系统依据已知蛋白质的pI值产生PI网络,使得凝胶上其它蛋白的PI按此分配。 所估计的精确度大大依赖于所建网格的结构及标本的类型。 已知的未被修饰的大蛋白应该作为标志,变性的修饰的蛋白的PI估计约在±0。25个单位。 同理,已知蛋白的理论分子量可以从数据库中计算,利用产生的表观分子量的网格来估计蛋白的分子量。 未被修饰的小蛋白的错误率大约30%,而翻译后蛋白的出入更大。 故需联合其他的技术完成鉴定。
传统的蛋白鉴定方法,如免疫印迹法、内肽的化学测序、已知或未知蛋白的comigration分析,或者在一个有机体中有意义的基因的过表达通常耗时、耗力,不适合高流通量的筛选。 目前,所选用的技术包括对于蛋白鉴定的图象分析、微量测序、进一步对肽片段进行鉴定的氨基酸组分分析和与质谱相关的技术。1 图象分析技术(Image analysis)“满天星”式的2-DE图谱分析不能依靠本能的直觉,每一个图象上斑点的上调、下调及出现、消失,都可能在生理和病理状态下产生,必须依靠计算机为基础的数据处理,进行定量分析。 在一系列高质量的2-DE凝胶产生(低背景染色,高度的重复性)的前提下,图象分析包括斑点检测、背景消减、斑点配比和数据库构建。 首先,采集图象通常所用的系统是电荷耦合CCD(charge coupled device)照相机;激光密度仪(laser densitometers)和Phospho或Fluoroimagers,对图象进行数字化。 并成为以象素(pixels)为基础的空间和网格。 其次,在图象灰度水平上过滤和变形,进行图象加工,以进行斑点检测。 利用Laplacian,Gaussian,DOG(difference of Gaussians) opreator使有意义的区域与背景分离,精确限定斑点的强度、面积、周长和方向。图象分析检测的斑点须与肉眼观测的斑点一致。 在这一原则下,多数系统以控制斑点的重心或最高峰来分析,边缘检测的软件可精确描述斑点外观,并进行边缘检测和邻近分析,以增加精确度。 通过阈值分析、边缘检测、销蚀和扩大斑点检测的基本工具还可恢复共迁移的斑点边界。 以PC机为基础的软件Phoretix-2D正挑战古老的Unix为基础的2-D分析软件包。 第三,一旦2-DE图象上的斑点被检测,许多图象需要分析比较、增加、消减或均值化。 由于在2-DE中出现100%的重复性是很困难的,由此凝胶间的蛋白质的配比对于图象分析系统是一个挑战。 IPG技术的出现已使斑点配比变得容易。 因此,较大程度的相似性可通过斑点配比向量算法在长度和平行度观测。 用来配比的著名软件系统包括Quest,Lips,Hermes,Gemini等,计算机方法如相似性、聚类分析、等级分类和主要因素分析已被采用,而神经网络、子波变换和实用分析在未来可被采用。 配比通常由一个人操作,其手工设定大约50个突出的斑点作为“路标”,进行交叉配比。 之后,扩展至整个胶。例如:精确的PI和MW(分子量)的估计通过参考图上20个或更多的已知蛋白所组成的标准曲线来计算未知蛋白的PI和MW。 在凝胶图象分析系统依据已知蛋白质的pI值产生PI网络,使得凝胶上其它蛋白的PI按此分配。 所估计的精确度大大依赖于所建网格的结构及标本的类型。 已知的未被修饰的大蛋白应该作为标志,变性的修饰的蛋白的PI估计约在±0。25个单位。 同理,已知蛋白的理论分子量可以从数据库中计算,利用产生的表观分子量的网格来估计蛋白的分子量。 未被修饰的小蛋白的错误率大约30%,而翻译后蛋白的出入更大。 故需联合其他的技术完成鉴定。2 微量测序(microsequencing)蛋白质的微量测序已成为蛋白质分析和鉴定的基石,可以提供足够的信息。 尽管氨基酸组分分析和肽质指纹谱(PMF)可鉴定由2-DE分离的蛋白,但最普通的N-末端Edman降解仍然是进行鉴定的主要技术。 目前已实现蛋白质微量测序的自动化。 首先使经凝胶分离的蛋白质直接印迹在PVDF膜或玻璃纤维膜上,染色、切割,然后直接置于测序仪中,可用于subpicomole水平的蛋白质的鉴定。 但有几点需注意:Edman降解很缓慢,序列以每40 min 1个氨基酸的速率产生;与质谱相比,Edman降解消耗大;试剂昂贵,每个氨基酸花费
SDS-PAGE测定乳基婴幼儿配方食品中乳清蛋白方法的研究
一、 前言基因工程已令人难以置信的扩展了我们关于有机体DNA序列的认识。但是仍有许多新识别的基因的功能还不知道,也不知道基因产物是如何相互作用从而产生活的有机体的。功能基因组试图通过大规模实验方法来回答这些问题。但由于仅从DNA序列尚不能回答某基因的表达时间、表达量、蛋白质翻译后加工和修饰的情况、以及它们的亚细胞分布等等,因此在整体水平上研究蛋白质表达及其功能变得日益显得重要。这些在基因组中不能解决的问题可望在蛋白质组研究中找到答案。蛋白质组研究的数据与基因组数据的整合,将会在后基因组研究中发挥重要作用。目前蛋白质组研究采用的主要技术是双向凝胶电泳和质谱方法。双向凝胶电泳的基本原理是蛋白质首先根据其等电点,第一向在pH梯度胶内等电聚焦,然后转90度按他们的分子量大小进行第二向的SDS-PAGE分离。质谱在90年代得到了长足的发展,生物质谱当上了主角,蛋白质组学又为生物质谱提供了一个大舞台。他们中首选的是MALDI-TOF,其分析容量大,单电荷为主的测定分子量高达30万,干扰因素少,适合蛋白质组的大规模分析。其次ESI为主的LC-MS联机适于精细的研究。本文将简介几种常用的生物质谱技术,并着重介绍生物质谱技术在蛋白质组学各领域的应用。二、 生物质谱技术1.电喷雾质谱技术(ESI)电喷雾质谱技术( Electrospray Ionization Mass Spectrometry , ESI - MS) 是在毛细管的出口处施加一高电压,所产生的高电场使从毛细管流出的液体雾化成细小的带电液滴,随着溶剂蒸发,液滴表面的电荷强度逐渐增大,最后液滴崩解为大量带一个或多个电荷的离子,致使分析物以单电荷或多电荷离子的形式进入气相。电喷雾离子化的特点是产生高电荷离子而不是碎片离子, 使质量电荷比(m/ z) 降低到多数质量分析仪器都可以检测的范围,因而大大扩展了分子量的分析范围,离子的真实分子质量也可以根据质荷比及电荷数算出。2.基质辅助激光解吸附质谱技术(MOLDI)基质辅助激光解析电离(MOLDI)是由德国科学家Karas和Hillenkamp发现的。将微量蛋白质与过量的小分子基体的混合液体点到样品靶上,经加热或风吹烘干形成共结晶,放入离子源内。当激光照射到靶点上时,基体吸收了激光的能力跃迁到激发态,导致蛋白质电离和汽化,电离的结果通常是基体的质子转移到蛋白质上。然后由高电压将电离的蛋白质从离子源转送到质量分析器内,再经离子检测器和数据处理得到质谱图。TOF质量分析器被认为是与MALDI的最佳搭配,因为二者都是脉冲工作方式,在质量分析过程中离子损失很少,可以获得很高的灵敏度。TOF质量分析器结果简单,容易换算,蛋白质离子在飞行管内的飞行速度仅与他的(m/z)-1/2成正比,因此容易通过计算蛋白质离子在飞行管内的飞行时间推算出蛋白质离子的m/z值。与传统质量分析器相比,更易得到高分辨率和高测量精度;速度快,离子飞行时间仅为几个μs和约100μs之间;质量范围宽,可以直接检测到几十万道尔顿的单电荷离子。飞行时间质量分析器被认为是21世纪最有应用前景的质量分析器。3.傅立叶变换-离子回旋共振质谱(FT-ICR MS)傅立叶变换-离子回旋共振质谱法(FT-ICR MS)是离子回旋共振波谱法与现代计算机技术相结合的产物。傅立叶变换-离子回旋共振质谱法是基于离子在均匀磁场中的回旋运动, 离子的回旋频率、半径、速度和能量是离子质量和离子电荷及磁场强度的函数, 当对离子施加与其回旋频率相同的射频场作用时, 离子将同相位加速到一较大的半径回旋, 从而产生可被接受的类似电流的信号。傅立叶变换-离子回旋共振质谱法所采用的射频范围覆盖了欲测定的质量范围,所有离子同时被激发, 所检测的信号经过傅立叶变换, 转换为质谱图。其主要优点有:容易获得高分辨;便于实现串极质谱分析;便于使用外电离源并与色谱仪器联用。此外,他还有灵敏度高,质量范围宽,速度快,性能可靠等优点。4.快原子轰击质谱技术(FABMS)快原子轰击质谱技术( Fast Atom Bomebardment Mass Spectrometry , FABMS) 是一种软电离技术,是用快速惰性原子射击存在于底物中的样品,使样品离子溅出进入分析器,这种软电离技术适于极性强、热不稳定的化合物的分析,特别适用于多肽和蛋白质等的分析研究。FABMS能提供有关离子的精确质量,从而可以确定样品的元素组成和分子式。而FABMS -MS 串联技术的应用可以提供样品较为详细的分子结构信息,从而使其在生物医学分析中迅速发展起来。三、蛋白质的分析鉴定随着质谱技术的发展,分子量的测定已从传统的有机小分子扩展到了生物大分子。MALDI-MS技术以其极高的灵敏度、精确度在蛋白质分析中得到了广泛的应用。该技术不仅可测定各种疏水性、亲水性和糖蛋白的分子量,还可直接测定蛋白质混合物的分子量。这可认为是蛋白质分析领域的一项重大突破。蛋白质组的研究是从整体水平上研究细胞或有机体内蛋白质的组成及其活动规律。质谱技术作为蛋白质组研究的三大支撑技术之一,除了用于多肽,蛋白质的分子量测定外,还广泛的应用于肽指纹图谱测定及氨基酸序列测定。肽指纹图谱(Peptide Mass Fingerprinting, PMF)测定是对蛋白酶解或降解后所得多肽混合物进行质谱分析的方法。质谱分析所得肽断与多肽蛋白数据库中蛋白质的理论肽断进行比较,判断出所测蛋白是已知还是未知。由于不同的蛋白质具有不同的氨基酸序列,不同蛋白质所得肽断具有指纹特征。采用肽指纹谱的方法已对酵母、大肠杆菌、人心肌等多种蛋白质组进行了研究。对肽序列的测定往往要应用串连质谱技术,采用不同的技术选择特定质核比的离子,并对其进行碰撞诱导解离,通过分析肽段的断裂情况推导出肽序列。四、后转录修饰的蛋白质的检测和识别在蛋白质组的研究中,蛋白质和多肽的序列分析已不局限于阐明蛋白质的一级结构,对翻译后的修饰的进一步分析也是蛋白质化学的一项重要任务。这种修饰对于蛋白质的功能非常重要,如:细胞识别中的蛋白质相互作用,信号传导和蛋白质定位。1. 蛋白质的糖基化糖蛋白在细胞内部,细胞膜和细胞外均有发现,实际上大部分蛋白质是糖蛋白。对糖蛋白的检测和分析发现,糖蛋白中糖组分的结构和功能具有多样性。糖蛋白中的糖通常是不同种类的,而且是由一些可控数量的单糖组成。糖基化的多样性与细胞周期,细胞分化和发展的状态有关。在蛋白组时代中,蛋白质的修饰会引起其理化性质的改变,因此是不容忽视的。从1D或2D凝胶得到的糖基化蛋白的识别,一般是进行MALDI-MS指纹分析, 或是对MALDI-PAD或ESI-MS/MS得到的碎片谱进行分析。对完整的糖蛋白的研究是非常困难的,所有已知的离子化技术都有其局限性。目前,人们主要研究糖肽,其好处之一就是质量减小了,这就会得到更好的分辨率,而且糖肽仍保留了糖基化位点。将分离的糖蛋白用不同的蛋白酶消化后就可进行糖肽的研究。一旦糖肽被识别出,就可以用串连质谱(ESI-MS/MS)来阐明肽序列。当蛋白的序列已知时,计算质量差就可推出其上附着的寡糖的质量。要将糖部分从糖蛋白中释放出来,可用化学切割或酶切割(流程图见图1)。目前,连有结构专一性糖苷酶的质谱在提供序列,分支和链接数据方面是最有力的技术。对于N糖基化常用的糖苷内切酶有PNGase-F, PNGase-A, EndoF和EndoH。化学切割也可以用来释放O-连接和N-连接的多糖,但经常出现的缺点是他会完全破坏所有的肽键,因而丢失了关于糖附着位点的信息。而且这些切割不能从糖肽中连续释放单糖。用肼的化学切割可以除去两种类型的糖基化。在60℃可专一性的释放O-连接的糖,而在95℃能释放N-连接的糖。释放O-原子更常用的方法是用碱进行β消除。通常,糖基中加入金属离子在MALDI和ESI中离子化。用MALDI-MS分析糖类的一个好的选择是将之与其他一些化合物混合,这样可以进一步提高灵敏度和分辨率。不同的质谱方法可以产生多糖的源后裂解(PSD)和碰撞诱导解离 (CID)谱,这可以给出有关糖的序列,分支及糖间的连接等信息。2. 蛋白质的磷酸化蛋白质中氨基酸的磷酸化在生命系统中起重要的作用。磷酸化经常作为分子开关控制不同过程蛋白质的活性,如新陈代谢,信号传导,细胞分裂等过程。因此,蛋白质中磷酰氨基酸的识别在蛋白质分析中是一项重要的工作。已知的磷酰氨基酸的类型有四种:1.O-磷酸盐,通过羟氨酸的磷酸化形成的,如丝氨酸,苏氨酸,酪氨酸。2.N-磷酸盐,通过精氨酸,赖氨酸或组氨酸中的氨基的磷酸化形成的。3.乙酰磷酸盐,通过天冬氨酸或谷氨酸的磷酸化形成的。4.S-磷酸酯,通过半胱氨酸的磷酸化形成的。
hey,大家好,不知道这里有没有蛋白组实验室的,想交流一下关于质谱性能测试样品的选择。我指的不是校正液哦。一般我们实验室就是平常自己BSA酶切后上质谱来看多肽峰的丰度,保留时间,当然搜库后还可以看看序列覆盖度等。有时评估一些前处理的技术有时也会用到BSA酶切后的多肽。我想问一下国内有没有卖已经酶切好的BSA?或者有没有比较便宜的TPCK-trypsin可以买? 老是用promega的质谱级胰酶切BSA感觉有点贵。谢谢了!
一、 前言 基因工程已令人难以置信的扩展了我们关于有机体DNA序列的认识。但是仍有许多新识别的基因的功能还不知道,也不知道基因产物是如何相互作用从而产生活的有机体的。功能基因组试图通过大规模实验方法来回答这些问题。但由于仅从DNA序列尚不能回答某基因的表达时间、表达量、蛋白质翻译后加工和修饰的情况、以及它们的亚细胞分布等等,因此在整体水平上研究蛋白质表达及其功能变得日益显得重要。这些在基因组中不能解决的问题可望在蛋白质组研究中找到答案。蛋白质组研究的数据与基因组数据的整合,将会在后基因组研究中发挥重要作用。 目前蛋白质组研究采用的主要技术是双向凝胶电泳和质谱方法。双向凝胶电泳的基本原理是蛋白质首先根据其等电点,第一向在pH梯度胶内等电聚焦,然后转90度按他们的分子量大小进行第二向的SDS-PAGE分离。质谱在90年代得到了长足的发展,生物质谱当上了主角,蛋白质组学又为生物质谱提供了一个大舞台。他们中首选的是MALDI-TOF,其分析容量大,单电荷为主的测定分子量高达30万,干扰因素少,适合蛋白质组的大规模分析。其次ESI为主的 LC-MS联机适于精细的研究。本文将简介几种常用的生物质谱技术,并着重介绍生物质谱技术在蛋白质组学各领域的应用。 二、 生物质谱技术 1.电喷雾质谱技术(ESI) 电喷雾质谱技术( Electrospray Ionization Mass Spectrometry , ESI - MS) 是在毛细管的出口处施加一高电压,所产生的高电场使从毛细管流出的液体雾化成细小的带电液滴,随着溶剂蒸发,液滴表面的电荷强度逐渐增大,最后液滴崩解为大量带一个或多个电荷的离子,致使分析物以单电荷或多电荷离子的形式进入气相。电喷雾离子化的特点是产生高电荷离子而不是碎片离子, 使质量电荷比(m/ z) 降低到多数质量分析仪器都可以检测的范围,因而大大扩展了分子量的分析范围,离子的真实分子质量也可以根据质荷比及电荷数算出。本帖与http://bbs.instrument.com.cn/shtml/20081130/1613156/重复,故锁帖!请版友在发帖前先搜索一下,以免出现重复贴。谢谢!斑竹您给的重复贴链接有误,并且我写贴前已经搜索了标题。SORRY,请LZ检索一下这个网址,开始那个是弄错了。
几种常用的蛋白鉴定方法传统的蛋白鉴定方法,如免疫印迹法、内肽的化学测序、已知或未知蛋白的comigration分析,或者在一个有机体中有意义的基因的过表达通常耗时、耗力,不适合高流通量的筛选。 目前,所选用的技术包括对于蛋白鉴定的图象分析、微量测序、进一步对肽片段进行鉴定的氨基酸组分分析和与质谱相关的技术。1 图象分析技术(Image analysis)“满天星”式的2-DE图谱分析不能依靠本能的直觉,每一个图象上斑点的上调、下调及出现、消失,都可能在生理和病理状态下产生,必须依靠计算机为基础的数据处理,进行定量分析。 在一系列高质量的2-DE凝胶产生(低背景染色,高度的重复性)的前提下,图象分析包括斑点检测、背景消减、斑点配比和数据库构建。 首先,采集图象通常所用的系统是电荷耦合CCD(charge coupled device)照相机;激光密度仪(laser densitometers)和Phospho或Fluoroimagers,对图象进行数字化。 并成为以象素(pixels)为基础的空间和网格。 其次,在图象灰度水平上过滤和变形,进行图象加工,以进行斑点检测。 利用Laplacian,Gaussian,DOG(difference of Gaussians) opreator使有意义的区域与背景分离,精确限定斑点的强度、面积、周长和方向。图象分析检测的斑点须与肉眼观测的斑点一致。 在这一原则下,多数系统以控制斑点的重心或最高峰来分析,边缘检测的软件可精确描述斑点外观,并进行边缘检测和邻近分析,以增加精确度。 通过阈值分析、边缘检测、销蚀和扩大斑点检测的基本工具还可恢复共迁移的斑点边界。 以PC机为基础的软件Phoretix-2D正挑战古老的Unix为基础的2-D分析软件包。 第三,一旦2-DE图象上的斑点被检测,许多图象需要分析比较、增加、消减或均值化。 由于在2-DE中出现100%的重复性是很困难的,由此凝胶间的蛋白质的配比对于图象分析系统是一个挑战。 IPG技术的出现已使斑点配比变得容易。 因此,较大程度的相似性可通过斑点配比向量算法在长度和平行度观测。 用来配比的著名软件系统包括Quest,Lips,Hermes,Gemini等,计算机方法如相似性、聚类分析、等级分类和主要因素分析已被采用,而神经网络、子波变换和实用分析在未来可被采用。 配比通常由一个人操作,其手工设定大约50个突出的斑点作为“路标”,进行交叉配比。 之后,扩展至整个胶。例如:精确的PI和MW(分子量)的估计通过参考图上20个或更多的已知蛋白所组成的标准曲线来计算未知蛋白的PI和MW。 在凝胶图象分析系统依据已知蛋白质的pI值产生PI网络,使得凝胶上其它蛋白的PI按此分配。 所估计的精确度大大依赖于所建网格的结构及标本的类型。 已知的未被修饰的大蛋白应该作为标志,变性的修饰的蛋白的PI估计约在±0。25个单位。 同理,已知蛋白的理论分子量可以从数据库中计算,利用产生的表观分子量的网格来估计蛋白的分子量。 未被修饰的小蛋白的错误率大约30%,而翻译后蛋白的出入更大。 故需联合其他的技术完成鉴定。2 微量测序(microsequencing)蛋白质的微量测序已成为蛋白质分析和鉴定的基石,可以提供足够的信息。 尽管氨基酸组分分析和肽质指纹谱(PMF)可鉴定由2-DE分离的蛋白,但最普通的N-末端Edman降解仍然是进行鉴定的主要技术。 目前已实现蛋白质微量测序的自动化。 首先使经凝胶分离的蛋白质直接印迹在PVDF膜或玻璃纤维膜上,染色、切割,然后直接置于测序仪中,可用于subpicomole水平的蛋白质的鉴定。 但有几点需注意:Edman降解很缓慢,序列以每40 min 1个氨基酸的速率产生;与质谱相比,Edman降解消耗大;试剂昂贵,每个氨基酸花费3~4$。 这都说明泛化的Edman降解蛋白质不适合分析成百上千的蛋白质。 然而,如果在一个凝胶上仅有几个有意义的蛋白质,或者如果其他技术无法测定而克隆其基因是必需的,则需要进行泛化的Edman降解测序。近来,应用自动化的Edman降解可产生短的N-末端序列标签,这是将质谱的序列标签概念用于Edman降解,业已成为一种强有力的蛋白质鉴定。 当对Edman的硬件进行简单改进,以迅速产生N-末端序列标签达10~20个/d,序列检签将适于在较小的蛋白质组中进行鉴定。若联合其他的蛋白质属性,如氨基酸组分分析、肽质质量、表现蛋白质分子量、等电点,可以更加可信地鉴定蛋白质。 选择BLAST程序,可与数据库相配比。 目前,采用一种Tagldent的检索程序,还可以进行种间比较鉴定,又提高了其在蛋白质组研究中的作用。3 与质谱(mass spectrometry)相关的技术质谱已成为连接蛋白质与基因的重要技术,开启了大规模自动化的蛋白质鉴定之门。 用来分析蛋白质或多肽的质谱有两个主要的部分,1)样品入机的离子源,2)测量被介入离子的分子量的装置。 首先是基质辅助激光解吸附电离飞行时间质谱(MALDI-TOF)为一脉冲式的离子化技术。 它从固相标本中产生离子,并在飞行管中测其分子量。 其次是电喷雾质谱(ESI-MS),是一连续离子化的方法,从液相中产生离子,联合四极质谱或在飞行时间检测器中测其分子量。 近年来,质谱的装置和技术有了长足的进展。在MALDI-TOF中,最重要的进步是离子反射器(ion reflectron)和延迟提取(delayed ion extraction),可达相当精确的分子量。 在ESI-MS中,纳米级电雾源(nano-electrospray source)的出现使得微升级的样品在30~40 min内分析成为可能。将反相液相色谱和串联质谱(tandem MS)联用,可在数十个picomole的水平检测;若利用毛细管色谱与串联质谱联用,则可在低picomole到高femtomole水平检测;当利用毛细管电泳与串联质谱连用时,可在小于femtomole的水平检测。 甚至可在attomole水平进行。 目前多为酶解、液相色谱分离、串联质谱及计算机算法的联合应用鉴定蛋白质。 下面以肽质指纹术和肽片段的测序来说明怎样通过质谱来鉴定蛋白质。(1)肽质指纹术(peptide mass fingerprint, PMF)由Henzel等人于1993年提出。 用酶(最常用的是胰酶)对由2-DE分离的蛋白在胶上或在膜上于精氨酸或赖氨酸的C-末端处进行断裂,断裂所产生的精确的分子量通过质谱来测量(MALDI-TOF-MS,或为ESI-MS),这一技术能够完成的肽质量可精确到0。1个分子量单位。 所有的肽质量最后与数据库中理论肽质量相配比(理论肽是由实验所用的酶来“断裂”蛋白所产生的)。 配比的结果是按照数据库中肽片段与未知蛋白共有的肽片段数目作一排行榜,“冠军”肽片段可能代表一个未知蛋白。若冠亚军之间的肽片段存在较大差异,且这个蛋白可与实验所示的肽片段覆盖良好,则说明正确鉴定的可能性较大。(2)肽片段(peptide fragment)的部分测序肽质指纹术对其自身而言,不能揭示所衍生的肽片段或蛋白质。 为进一步鉴定蛋白质,出现了一系列的质谱方法用来描述肽片段。 用酶或化学方法从N-或C-末端按顺序除去氨基酸,形成梯形肽片段(ladder peptide)。 首先以一种可控制的化学模式从N-末端降解,可产生大小不同的一系列的梯形肽片段,所得一定数目的肽质量由MALDI-TOF-MS测量。 另一种方法涉及羧基肽酶的应用,从C-末端除去不同数目的氨基酸形成肽片段。 化学法和酶法可产生相对较长的序列,其分子量精确至以区别赖氨酸(128。09)和谷氨酰胺(128。06)。 或者,在质谱仪内应用源后衰变(post-source decay, PSD)和碰撞诱导解离(collision-induced dissociation, CID),目的是产生包含有仅异于一个氨基酸残基质量的一系列肽峰的质谱。 因此,允许推断肽片段序列。 肽片段PSD的分析在MALDI反应器上能产生部分序列信息。 首先进行肽质指纹鉴定。 之后,一个有意义的肽片段在质谱仪被选作“母离子”,在飞行至离子反应器的过程中降解为“子离子”。 在反应器中,用逐渐降低的电压可测量至检测器的不同大小的片段。 但经常产生不完全的片段。 现在用肽片段来测序的方法始于70年代末的CID,可以一个三联四极质谱ESI-MS或MALDI-TOF-MS联合碰撞器内来完成。 在ESI-MS中,由电雾源产生的肽离子在质谱仪的第一个四极质谱中测量,有意义的肽片段被送至第二个四极质谱中,惰性气体轰击使其成为碎片,所得产物在第三个四极质谱中测量。 与MALDI-PSD相比,CID稳定、强健、普遍,肽离子片段基本沿着酰胺键的主架被轰击产生梯形序列。 连续的片段间差异决定此序列在那一点的氨基酸的质量。 由此,序列可被推测。 由CID图谱还可获得的几个序列的残基,叫做“肽序列标签”。 这样,联合肽片段母离子的分子量和肽片段距N- C端的距离将足以鉴定一个蛋白质。4 氨基酸组分分析1977年首次作为鉴定蛋白质的一种工具,是一种独特的“脚印”技术。 利用蛋白质异质性的氨基酸组分特征,成为一种独立于序列的属性,不同于肽质量或序列标签。 Latter首次表明氨基酸组分的数据能用于从2-DE凝胶上鉴定蛋白质。 通过放射标记的氨基酸来测定蛋白质的组分,或者将蛋白质印迹到PVDF膜上,在15







 我要推广仪器
我要推广仪器
 下载APP
下载APP