本人刚接触液相色谱,想要检测某饮料中某一物质的含量采用外标法,该物质的大体含量不清楚,看了几篇参考文献说外标法中浓度范围最好包括样品浓度,这该怎么做呢?难道扩大外标法中的浓度范围吗,这样好像比较麻烦又不见得准确啊?还有就是进样量应该怎么确定啊?
[color=#444444]液相色谱使用外标法定量,但是经常出现所有组分含量总和超过100%的现象~~这是为什么,希望高人能够指点一下。[/color]
岛津的液相色谱,配有自动进样器。用外标定量时,只配一个标样,选择用改变的进样量(即1微升,2微升。。。。)这种方式来做标准曲线,是不是线性更好一些。
高效[url=https://insevent.instrument.com.cn/t/5p][color=#3333ff]液相色谱[/color][/url](HPLC)中外标法是一种常用的定量分析方法,其基本概念是通过测量已知浓度的标样(外标)和未知浓度的样品的色谱峰面积或峰高,从而计算出样品中目标物质的浓度。一般分为外标单点法和外标曲线法,现在对不同国家的外标法进行对比。 [size=18px][b][color=#595959]01.中国药典中的外标法(2025版的公示稿) [/color][/b][/size][b][size=16px][color=#595959]1)外标法的一般步骤:[/color][/size][size=16px][color=#ff4c41](可以理解为外标法) [/color][/size][/b][size=16px][color=#595959]a.按照各品种项下的规定,精确称取或量取对照品和供试品,配制成溶液; [/color][/size][size=16px][color=#595959]b.分别精确量取对照品溶液和供试品溶液的一定体积,进行色谱分析; [/color][/size][size=16px][color=#595959]c.记录色谱图,并测量待测物质的峰面积(或峰高); [/color][/size][size=16px][color=#595959]d.使用公式 C[sub]X[/sub]=C[sub]R[/sub]×A[sub]X[/sub]/A[sub]R[/sub]计算含量,其中 C[sub]X[/sub]是供试品中待测物质的浓度,C[sub]R[/sub]是对照品的浓度,A[sub]X[/sub]是供试品溶液中待测物质的峰面积(或峰高),A[sub]R[/sub]是对照品溶液中待测物质的峰面积(或峰高)。 [/color][/size][b][size=16px][color=#595959]2)[/color][/size][size=16px][color=#595959]对于成分复杂或待测成分含量变化较大的药物体系,可以采用[/color][/size][size=16px][color=#ff4c41]外标标准曲线法[/color][/size][size=16px][color=#595959]进行含量测定。[/color][/size][/b][size=16px][color=#595959]a.精确称取适量的对照品,或从对照品储备液中精确量取,配制成不同浓度的系列溶液; [/color][/size][size=16px][color=#595959]b.精确量取这些系列溶液,进行色谱分析,记录色谱图,并测量峰响应; [/color][/size][size=16px][color=#595959]c.利用峰响应(或经转换)对浓度绘制标准曲线,并通过最小二乘法计算出回归曲线方程; [/color][/size][size=16px][color=#595959]d.在相同的色谱条件下,精确量取供试品溶液,进行色谱分析,记录色谱图,并测量待测成分的峰响应; e[/color][/size][size=16px][color=#595959].根据待测成分的峰响应(或经转换)和回归曲线方程,确定供试品溶液中待测成分的含量。[/color][/size][size=16px][color=#ff4c41]简单理解就是做一条标准曲线,得出标准曲线方程,再将待测品的峰相应代入方程进行计算,得出样品浓度。 [/color][/size][size=16px][color=#ff4c41][b][color=#595959]02.欧洲药典中的外标法 [/color][/b][/color][/size][color=#ff4c41][b][size=16px][color=#595959]1)[b]多点校准(使用校准函数): [/b][/color][/size][/b][/color][size=16px][color=#595959]a.准备一系列含有不同梯度浓度的对照品溶液,这些浓度应该在已经证明可以得到线性响应的范围内; [/color][/size][size=16px][color=#595959]b.取固定体积的这些对照品溶液进行色谱分析; [/color][/size][size=16px][color=#595959]c.通过色谱图得到峰面积或峰高,并以此绘制校正曲线,横坐标是对照品的量,纵坐标是峰面积或峰高; [/color][/size][size=16px][color=#595959]d.通常通过线性回归得到校准函数; [/color][/size][size=16px][color=#595959]e.按照各论中规定的程序准备待测样品溶液; [/color][/size][size=16px][color=#595959]f.在与制备校准函数相同的操作条件下进行色谱分析; [/color][/size][size=16px][color=#595959]g.测量待测组分的峰面积或峰高,并根据校准函数读取或计算组分的含量; [/color][/size][size=16px][color=#595959]2[b])单点校准: [/b][/color][/size][size=16px][color=#595959]a.在各论中,通常选择一个校准曲线线性范围内的对照品溶液,以及一个浓度接近该对照品溶液的待测样品溶液; [/color][/size][size=16px][color=#595959]b.在固定条件下进行色谱分析,通过比较得到的响应来定量分析组分的含量; [/color][/size][color=#595959]c.在这种方法中,所有操作步骤,如进样程序,都必须在恒定条件下进行 [img=,690,801]https://ng1.17img.cn/bbsfiles/images/2024/09/202409061421193322_2231_3203140_3.jpg!w690x801.jpg[/img] 图1.欧洲药典原文描述[b][color=#595959]03.美国药典中的外标法 [/color][/b][size=16px][color=#595959][b]1)校准函数法(多点校准): [/b][/color][/size][size=16px][color=#595959]a.制备一系列含有不同浓度的对照品标准溶液,这些浓度应在已知能够产生线性响应的范围内; b[/color][/size][size=16px][color=#595959].取固定体积的这些标准溶液进行色谱分析,并注射到色谱系统中; [/color][/size][size=16px][color=#595959]c.通过色谱图得到的峰面积或峰高,绘制校准曲线,横坐标是对照品的量,纵坐标是峰面积或峰高; [/color][/size][size=16px][color=#595959]d.通常通过线性回归方法得到校准函数; [/color][/size][size=16px][color=#595959]e.根据各论中规定的程序制备待测样品溶液; [/color][/size][size=16px][color=#595959]f.在与制备校准函数相同的色谱条件下分析样品,测量待测组分的峰面积或峰高; [/color][/size][size=16px][color=#595959]g.通过校准函数计算出样品中待测组分的含量。[/color][/size][size=16px][color=#595959][b]2)单点校准法:[/b][/color][/size][size=16px][color=#595959]a.在各论中,选择一个浓度在线性范围内的标准溶液,以及一个浓度接近该标准溶液的待测样品溶液; [/color][/size][size=16px][color=#595959]b.在固定条件下进行色谱分析,通过比较得到的响应来定量分析组分的含量; [/color][/size][color=#595959]c.这种方法中,所有操作步骤,包括进样程序,都必须在恒定条件下进行,以确保分析结果的一致性 [img=,690,214]https://ng1.17img.cn/bbsfiles/images/2024/09/202409061424187996_390_3203140_3.jpg!w690x214.jpg[/img] 图2.美国药典原文描述 [color=#595959][b]总结一下:[/b][/color][size=16px]1)中国药典提供了外标法的一般步骤和标准曲线法的详细描述,但没有明确描述单点校准法。 [/size][size=16px]2)欧洲药典和美国药典都详细描述了多点校准和单点校准的方法,并且强调了所有操作步骤需要在恒定条件下进行。(这两家基本是一 致的描述) [/size][size=16px]3)三种药典都强调了在色谱分析中保持操作条件的一致性,以确保分析结果的准确性和重现性 [img=,690,249]https://ng1.17img.cn/bbsfiles/images/2024/09/202409061426098030_8014_3203140_3.jpg!w690x249.jpg[/img] [/size][/color][/color]
液相色谱和[url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]的外标法和内标定量有何区别?是如何处理的?
我想问下,外标法做标准曲线的时候,进样量是不是要精确啊,液相色谱有定量环可以精确定量,[url=https://insevent.instrument.com.cn/t/Mp]气相[/url]没有,那是不是就是根据注射器体积定进样量啊,如果是自动进样的话会比较精确,如果是手动的话 那是不是就会不准了?
请问各位专家:液相色谱外标法测定含量(80%左右)时一般允许的误差是多少呢?2%以内可以吗?
做液相色谱正相外标的时候,如果样品峰面积和稀释100倍后的对照液的峰面积不成100:1的时候该怎么办?
[color=#444444]最近做的不同批次的同一样品,用的是安捷伦1200液相色谱仪,流动相为70%乙腈,波长为215nm。结果如下[/color][color=#444444]1、用面积归一化法计算,纯度为85%,可是用外标法计算含量时,却高达97%(没有乘以总重量,下同)[/color][color=#444444]2、另测一批样品,归一化纯度为86%,外标法计算含量时,却高达100%。[/color][color=#444444]请问这是什么情况?[/color]
液相色谱,外标法定量,对标准样品的纯度有要求吗?是不是一定要大于98%?现在只能买到FLUKA质保书上纯度为95%的标准品(GC),可以用于外标法定量吗。大家平时有没有碰到过标准品纯度不高的问题,是不是说明这种物质很难提纯?
如题,外标法在液相色谱中是使用最广的。但由于[url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]的进样重复性没有液相色谱那么理想,使用内标法比较多,但现在仪器技术发展了,用外标法是主流了吗?
各位好,我刚学习液相,不是很明白在做液相时内标法和外标法,哪位能详细解释下啊。谢谢~~~
使用液相色谱定量分析时候采用外标法分析,请问按国家标准的话,配制标准溶液和试样溶液各几份?目前我都是配一个标准溶液 和2份 试样溶液(标识:试样A+试样B)分析的,请问这可行吗?进样顺序如下:进数针标样平行后+ 2针试样A + 2针试样B + 1针标样 请问下同行了兄弟姐妹,这样做法可行否?还有算结果时我是取最后1针标样和数针标样中的一针加和平均来算
[color=#444444]请问,液相色谱测定含量双样双针的,f值有没有统一规定的啊?F=称样量/A峰面积/稀释倍数,要不要除以对照品的稀释体积啊?是随便写还是有统一规定写法啊?[/color]
用高效液相色谱仪外标法测定兽药含量可不可以不建立标准曲线?测出标准品和样品的色谱峰之后直接带入公式求含量可以吗?
想问一下,一般大家测定一种化合物时,是自建方法还是参照国标呢?用HPLC定量时,如果使用外标法,那么多久需要重新做一条标准曲线呢?还是每次测定样品的时候,都需要同时测定标准品,绘制新的标准曲线?在做标准曲线的时候,是配制不同浓度的标准品(比如1mg/25ml,2mg/25ml,4mg/25ml,8mg/25ml,10mg/25ml...),然后进相同的进样量(比如10ul),还是配制1个浓度的标准品然后进不同进样量?
同一个样品,检测两次,均采用外标法,一个是气相色谱,一个是液相色谱含量差距很多,气相含量是百分之90左右,液相含量是百分之95左右,求大神分析原因?
大家好,请大家来帮助讨论一下外标法测一样品的杂质含量:我现在有一个样品要求用外标法测其含量小于100ppm,现在已经有一杂质的标准品,请问如用外标法测的话如何配制标准杂质的浓度和样品浓度?
使用液相色谱定量分析时候,外标法。配制标准溶液和试样溶液各几份?有没必要配制2标样2试样?我通常一份标准,一份试样溶液,待标样溶液进样相对标准偏差在1.5%以内,按标样,试样,试样,标样的顺序做的。请大家说说你们通常是怎么做的?
液相色谱法检测含量是液相分析工作的重要的一项,液相色谱法检测含量用的基本是外标法和内标法。其中外标法检测的准确性受到更多因素的影响,更需要丰富的经验来避免或减小误差。本人是做农药制剂分析出身,经过多年的锻炼,自认为对外标法含量检测小有心得,加之最近论坛有坛友问及相关问题,所以想写一篇这方面的小文章,以供大家参考,同时也希望大家补漏拾遗,如有不对的地方,也请指出。首先,我们来看看外标法含量检测的计算公式:含量=m对*P*A样*V样/(m样*A对*V对)*100%其中的各个符号的意义大家都知道,我就不赘述。这其中的任何一项发生了改变,都会使得检测结果发生变化,而这其中的任何一项,都有很多种原因使其发生变化。下面我们就逐一分析。1. m对/m样——对照品称样量/样品称样量1.1 天平准确性天平是否经过校准,其是否在正常工作状态,称量时天平是否带有静电,是否有空气流动,这些都是影响读数的因素。1.2 称量方式采用的是加量法还是减量法,都是很有讲究的。正常情况下,肯定要采用加量法,一次称取到位,这样才能得到准确的称样量。1.3 称量操作称量时,是否足够小心,不会在转移样品时使样品撒漏,这个是非常重要的。1.4 样品性质样品如果具有挥发性,直接称样时,必然会挥发一部分造成误差;另外,如果样品不稳定,或在溶液中不稳定,那么实际上来讲,也是影响了样品量。还有,样品要保证均匀,固体不必说,液体样品要保证是均相,称量之前要摇匀。1.5 称样量对于万分之一天平,称样量最好能在100mg以上;对于十万分之一的天平,称样量应在10mg以上。 2. P——对照品含量2.1 对照品质量对照品是否有合法来源,其含量值是否准确,这个相信大家都知道。2.2 对照品的保存对照品是否按要求保存,也是至关重要的。因为如果对照品没有按照规定保存,那么其右可能会发生降解,进而影响含量值;另外,如果对照品是保存在冰箱中,称量时,应取出放在干燥器中回至室温再称量,否则其很有可能会吸收空气中的水分,影响含量值。同理,如果对照品引湿性很强的话,长期暴露在空气中,也会使其P发生变化。 3. A样/A对——样品峰面积/对照品峰面积3.1 方法的可靠性如果所用的分析方法可靠性不好,样品峰面积或保留时间容易变化,那么检测结果也是很重现性不好的。关于方法的可靠性,做过分析方法验证的人应该都能明白,这里包括系统适用性、精密度、准确性和耐用性以及线性等。3.2 仪器进样精密度这其实可以包括在上述3.1因素中。3.3 残留或污染如果仪器系统被污染产生残留,而本身峰面积就不大的情况下,这个因素产生的误差就更明显了。3.4 积分如果样品峰形不够好,或者分离度不够好的情况下,积分就很重要。 4. V样/V对——样品稀释体积/对照品稀释体积4.1 容量瓶、移液管等体积是否准确一般来说,新买的容量瓶体积是符合要求的,但是也难免会买到个别次品,这就需要进行校验了。4.2 容量瓶、移液管规格这个是很重要的,一般大家都能注意这个问题,做到合理使用。4.3 容量瓶等使用习惯容量瓶在使用时,不能使其至于高于40摄氏度的环境下,否则容量瓶会发生变形。我们以前有个习惯,冬天尤其频繁——瓶子不够用了,而洗过的还没干,就用电吹风吹干,这种行为不光会影响体积,还会使容量瓶瓶口发生变化,摇匀时造成漏液现象。4.4 定量操作这个包括容量瓶和移液管的使用,正确的使用方法就不用说了吧。我只说一些注意事项,一是移液管吸液时,液面不要高刻度太多,放液时千万不要用洗耳球吹,并且尽量保持每次操作时间都一样,粘度大的溶液尤其需要如此。二是观察液面时,要使容量瓶和移液管保持垂直,这时很多人喜欢用三根手指捏住,其实这样不容易保持垂直,最好用两根手指,轻轻捏住就好。三是注意接触容量瓶移液管等的面积越小越好,以免体温加热溶液。4.5 温度[font=Times New Roma
有做聚乙氧基化非离子表面活性剂中聚乙二醇含量的测定 高效液相色谱法 的没有,请教下外标法做曲线,聚乙二醇标样600-2000,是只取一种配制不同浓度,做曲线,还是600,1000,2000的都取。具体怎么个情况?
我们开展新的产品的检测方法,1.3环己二酮的含量检测,用乙腈和0.5%乙酸水做流动相(25:75),254波长紫外检测器。用的是单标的外标法检测,取样0.02g,定容50mL,然后稀释后进样的进样浓度(40ppb左右),然后我们测得样品含量的数据很不稳定,前天95-97%左右,昨天99%-103%,今天还是101%左右。求高手指导改进什么地方!!!注:1,单个浓度点的样品的峰面积重现性很好标准偏差0.4%左右。2,我们样品含量应该不超100%,用单标测别家的产品在99%左右。
请教高效液相色谱仪在进行含量测定时相对偏差为多少?对照品和供试品应各配几份,各进几针平行样?用外标法怎样计算? 谢谢!
[color=#444444]如果用液相色谱,GPC软件,要测定异戊橡胶的分子量,根据什么选择毛细管柱呢?请详细回答。[/color][color=#444444]用[url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]测定水中的微量乙腈,用外标法,为什么要用峰面积定量,用峰面积百分比可以吗?因为我们是手动进样器。请回答一下。谢谢[/color]
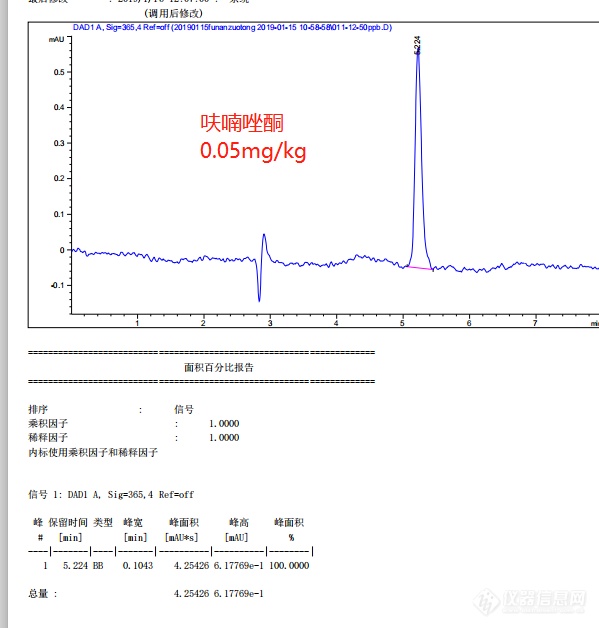
之前比较少做液相的实验,想让各位前辈指点一下。检测依据是SCT 3022-2004 水产品中呋喃唑酮残留量的测定液相色谱法,用峰高计算,外标法定量
液相色谱制作校正曲线以后,使用曲线时,打印报告结果显示是实际值的0.1倍
药典液相色谱方法的调整 • 根据药典附录“液相色谱法”规定,可调整适当参数 ---调整目的:满足系统适用性的要求 ---系统适用性的要求 ---HPLC方法调整的考虑因素 05版药典的系统适用性要求1、理论塔板数: ----反映整个色谱系统的状态 填料状态 管线连接 ----有不同的计算方法 主要是峰宽取值方法不同 不同计算方法计算结果有差异 ----影响因素: 被测组分的保留时间、进样量等 积分参数 系统死体积 测定色谱方法、样品与计算方法保持恒定,以便比较 05版药典的系统适用性要求 2、分离度: ---影响因素: * 影响柱效的因素 色谱柱尺寸 填料性能 进样量 * 影响分离选择性的因素 流动相组成 色谱柱品牌 柱温 * 柱外体积 ---有不同的计算方法,结果有差异 05版药典的系统适用性要求3、重复性(进样精密度): * 外标法:对照品溶液(n:5) 峰面积RSD:2.0% * 内标法:相当于80%,100%,120%的对照品溶液,加入规定量内标 溶液,分别至少进样2次,计算平均校正因子([font=Times New Roman
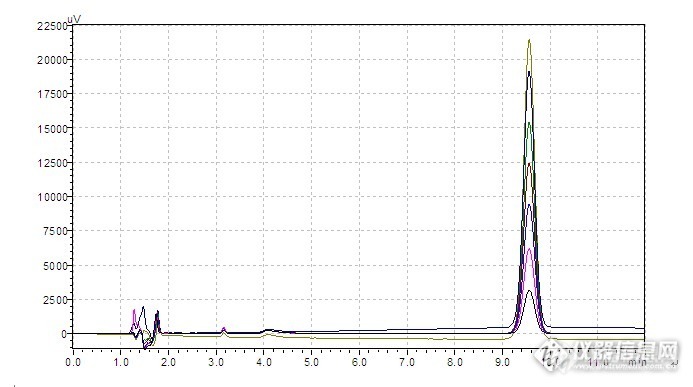
前言:前段时间药典委员会培训时一个老师讲过,以后,线性关系试验也可以用不同的进样量来进行。我想了想,这样做真好,省事多了啊!于是,来验证一下是否可行。溶出度试验条件:色谱条件与系统适用性以十八烷基硅烷键合硅胶为填充剂;以乙腈-0.01mol/L磷酸二氢钾溶液(用磷酸调节pH值至3.0)(45:55)为流动相;检测波长210 nm理论板数以****峰计算应不低于2000。取本品,照溶出度测定法(中国药典2010年版二部附录X C第二法),以水900ml为溶剂,转速为每分钟50转,依法操作,经15分钟时,取溶液10ml,滤过,取续滤液作为供试品溶液;另取****对照品10mg,精密称定,置100ml量瓶中,用甲醇溶解并稀释至刻度,摇匀,精密量取5ml,用水稀释至100ml,作为对照品溶液。取上述两种溶液各20μl,注入液相色谱仪,记录色谱图,按外标法以峰面积计算溶出量。限度为标示量的85%,应符合规定。色谱柱信息:welchrom C18 4.6*150mm,5微米PN:WEL 518415;SN:W11212195;LN:W1811.02初步情况详见图:http://ng1.17img.cn/bbsfiles/images/2012/12/201212010018_408484_1621890_3.jpg 进样量:20微升线性关系:http://ng1.17img.cn/bbsfiles/images/2012/12/201212010048_408488_1621890_3.bmp上面色谱图为20微升进样色谱图,下图为体积进样色谱图,两者的进样量一样,从色谱图上发现基本上无明显差异。http://ng1.17img.cn/bbsfiles/images/2012/12/201212010052_408489_1621890_3.bmphttp://ng1.17img.cn/bbsfiles/images/2012/12/201212010041_408485_1621890_3.gifhttp://ng1.17img.cn/bbsfiles/images/2012/12/201212010044_408487_1621890_3.gif试验总结:从以上图谱对比,由于进样量的控制主要由自动进样器来操作的,由于不同的进样量,其溶剂峰也是有一定的差异的,详见最后几张张图对比图可以看出。有些品种,用这种方法也可能会不成功的,主要是扩散效应是否可以忽略。
药典液相色谱方法的调整 • 根据药典附录“液相色谱法”规定,可调整适当参数 ---调整目的:满足系统适用性的要求 ---系统适用性的要求 ---HPLC方法调整的考虑因素 05版药典的系统适用性要求1、理论塔板数: ----反映整个色谱系统的状态 填料状态 管线连接 ----有不同的计算方法 主要是峰宽取值方法不同 不同计算方法计算结果有差异 ----影响因素: 被测组分的保留时间、进样量等 积分参数 系统死体积 测定色谱方法、样品与计算方法保持恒定,以便比较 05版药典的系统适用性要求 2、分离度: ---影响因素: * 影响柱效的因素 色谱柱尺寸 填料性能 进样量 * 影响分离选择性的因素 流动相组成 色谱柱品牌 柱温 * 柱外体积 ---有不同的计算方法,结果有差异 05版药典的系统适用性要求3、重复性(进样精密度): * 外标法:对照品溶液(n:5) 峰面积RSD:2.0% * 内标法:相当于80%,100%,120%的对照品溶液,加入规定量内标 溶液,分别至少进样2次,计算平均校正因子([font=Times New Roman
[color=#444444]如题[/color][color=#444444]我是用[url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]外标法 现在做工作曲线[/color][color=#444444]配置不同质量分数的A溶液 现在发现(以10%,wt 为例)同样的进样量 A物质出峰的面积相差悬殊(远在进样误差之外)有时候是1000+ 有时候是3000+[/color][color=#444444]请问大神是什么原因?[/color]







 我要推广仪器
我要推广仪器
 下载APP
下载APP