[b]超高效液相色谱串联质谱法是什么方法[/b]
[b][size=20px][color=#333333]丹磺酰氯柱前衍生-超高效液相色谱-串联质谱法测定人体尿样中的环己胺[/color][/size][/b]
高效液相色谱串联质谱可以定量吗?
[b]高效液相色谱三重四极杆串联质谱联用仪可以进行哪些工作模式?以及采用这些工作模式有什么作用?[/b]

[align=center][font='times new roman'][size=16px]超高效[url=https://insevent.instrument.com.cn/t/5p][color=#3333ff]液相色谱[/color][/url]四极杆飞行时间串联质谱仪[/size][/font][font='times new roman'][size=16px]的应用[/size][/font][/align][align=left][/align][font='宋体'][size=16px]中广测配备了超高效[url=https://insevent.instrument.com.cn/t/5p][color=#3333ff]液相色谱[/color][/url]四极杆飞行时间串联质谱联用仪(UPLC-Q-TOF/MS),具有分析速度快、质量精度高、重复性好等优点,可以提供精确分子量和分子式等信息,是目前未知物成分鉴定和化学成分定性定量分析的主要设备之一,在日用化工等各领域的成分分析鉴定中已有广泛应用。[/size][/font][align=center][font='宋体'][size=16px][color=#444444] [/color][/size][/font][img]https://ng1.17img.cn/bbsfiles/images/2023/10/202310271103180399_1739_2862401_3.png[/img][/align][font='宋体'][size=16px]一、仪器信息[/size][/font][font='宋体'][size=16px]1.仪器名称:超高效[url=https://insevent.instrument.com.cn/t/5p][color=#3333ff]液相色谱[/color][/url]-四极杆飞行时间质谱联用仪[/size][/font][font='宋体'][size=16px]2.英文名称:UPLC-Q-TOF/MS[/size][/font][font='宋体'][size=16px]3.生产制造商:Agilent Technologies Co., Ltd.[/size][/font][font='宋体'][size=16px]4.型号:Agilent 1290UHPLC&6540B QTOF-MS[/size][/font][font='宋体'][size=16px]二、主要技术指标[/size][/font][font='宋体'][size=16px]1.质量范围:飞行时间质谱(TOF)的质量范围m/z 25~20000;[/size][/font][font='宋体'][size=16px]2.灵敏度:2pg的利血平在ESI一级质谱全扫描(TOF-MS)模式下的信噪比(S/N)不低于10:1,二级质谱(Q-TOF-MS/MS)模式下的S/N不低于50:1;[/size][/font][font='宋体'][size=16px]3.质量精度:100pg的利血平(Reserpine)在一级质谱全扫描模式下获得的母离子m/z 609.28066的质量偏差<1ppm,其二级质谱的特征碎片离子m/z 397.21218的质量偏差<2ppm;[/size][/font][font='宋体'][size=16px]4.分辨率:m/z 1521.971475离子的分辨率>40000。[/size][/font][font='宋体'][size=16px]三、应用领域[/size][/font][font='宋体'][size=16px]新药研发、药物代谢、有机化合物定性、环境分析、法医毒物、残留分析、食品安全、产品检验、卫生防疫等领域。[/size][/font][font='宋体'][size=16px]四、服务范围[/size][/font][font='宋体'][size=16px]1.精确分子量测试:可为高等院校、科研院所及各类企业提供化合物准确分子测试服务;[/size][/font][font='宋体'][size=16px]2.药物有关物质定性分析:可为企业或研究机构提供药品中有关物质的高分辨质谱定性分析;[/size][/font][font='宋体'][size=16px]3.药物代谢研究:新药开发的临床研究,药代动力学研究,生物利用度研究,代谢产物的研究等;[/size][/font][font='宋体'][size=16px]4.化学应急毒物分析:利用该仪器高分辨、高质量精度、高灵敏度的特点,可为化学中毒应急突发事件提供准确可靠的定性结果,为应急预案、应急处理提供科学依据;[/size][/font][font='宋体'][size=16px]5.未知有机化合物的研究:利用仪器对精确分子量的测定,可提供分子式及同位素信息的功能,分析鉴定未知有机化合物的化学结构。[/size][/font][font='宋体'][size=16px]五、应用案例[/size][/font][font='宋体'][size=16px]1.中药物质基础研究[/size][/font][font='宋体'][size=16px]中药物质基础研究是阐明中药药效物质、药理作用及其机制和临床疗效的先决条件,也是深层次开发中药方剂、改进工艺和剂型、制定质量标准、提高临床疗效的重要基础,是中药现代化的重要组成部分。中广测利用HPLC-Q-TOF/MS技术,结合一级准确质量数及二级碎片离子信息,对数十种中药或复方的物质基础进行了研究,并建立了中药化学成分数据库。目前已鉴定的中药包括:沉香、广陈皮、广藿香、佛手、阳春砂、何首乌、巴戟天、化橘红、甘草、茯苓、火炭母、吴茱萸、苦杏仁、桃仁等数十种中药材及中药复方。[/size][/font][font='宋体'][size=16px][color=#444444] [/color][/size][/font][img]https://ng1.17img.cn/bbsfiles/images/2023/10/202310271103183069_495_2862401_3.png[/img][align=left][/align][font='宋体'][size=16px]2.植物代谢组学研究[/size][/font][font='宋体'][size=16px]不同产地、不同种类或不同炮制方法的中药化学成分的差异是中药药效研究或质量控制的关键。中广测利用HPLC-Q-TOF/MS技术,结合植物代谢组学与统计分析方法实现了不同产地中药材、不同类型中药材的鉴定,以及中药炮制前后化学成分的变化。如广东、广西与湖南三地的陈皮的区分,苦杏仁与桃仁化学成分的差异以及广佛手蒸制前后化学成分的变化。 [/size][/font][align=center][/align][align=center][img]https://ng1.17img.cn/bbsfiles/images/2023/10/202310271103187647_7125_2862401_3.png[/img][/align][align=left][/align][align=center][img]https://ng1.17img.cn/bbsfiles/images/2023/10/202310271103188224_1520_2862401_3.png[/img][/align][align=left][/align][font='宋体'][size=16px]3.西药杂质定性分析[/size][/font][font='宋体'][size=16px]杂质的研究是药品开发的一项重要内容。杂质是否能被全面准确地加以控制,直接关系到药品的质量可控性与安全性。因此,在药物的研究、生产、供应和临床使用等方面,必须保证药物的纯度,规范地进行杂质的研究。利用HPLC-Q-TOF/MS技术可通过化合物精确质量数得到其分子式,结合主成分及杂质的二级质谱信息以及药物合成路线,推测出杂质结构及其来源。[/size][/font][align=center][img]https://ng1.17img.cn/bbsfiles/images/2023/10/202310271103191461_9895_2862401_3.png[/img][/align]
超高效液相色谱-串联质谱检测动物源性食品中β-受体激动剂残留分析方法的优化
:高效液相色谱_串联质谱法测定酸奶中纳他霉素的含量期刊:中国卫生检验杂志作者:李媛媛; 王伟; 关树文; 李刚; 董巧红;链接:http://202.119.208.220:8002/kns50/detail.aspx?dbname=CJFD2010&filename=ZWJZ201004027
固相萃取-超高效[url=https://insevent.instrument.com.cn/t/5p][color=#3333ff]液相色谱[/color][/url]法-串联质谱法测定蜂蜜中甲硝唑[color=black] [/color][font=宋体][color=black]甲硝唑(MNZ)属于硝基咪唑类广谱抗生素,广泛用于预防和治疗组织滴虫病、球虫病等疾病,甲硝唑因疗效明显,价格低廉,被蜂农广泛使用,造成了甲硝唑药物在蜂蜜中残留[/color][/font][font=宋体][sup][size=13px][1,2][/size][/sup][/font][font=宋体][color=black],研究发现甲硝唑对人体具有潜在的致癌和致畸作用[/color][/font][font=宋体][sup][size=13px][3,4][/size][/sup][/font][font=宋体][color=black]。1998年欧盟禁止甲硝唑使用于食品动物,2002年美国食品与药物监督管理局禁止在进口动物源性食品中使用甲硝唑[/color][/font][font=宋体][sup][size=13px][5,6][/size][/sup][/font][font=宋体][color=black]。我国农业部和国家药品监督管理局2002年规定甲硝唑及其盐、酯及制剂不准以促进动物生长为目的在所有食品动物饲养过程中使用,且不得在动物源食品中检出[/color][/font][font=宋体][sup][size=13px][7,8][/size][/sup][/font][font=宋体][color=black]。目前甲硝唑的测定方法主要有[url=https://insevent.instrument.com.cn/t/Mp][color=#3333ff]气相色谱[/color][/url]法、高效[url=https://insevent.instrument.com.cn/t/5p][color=#3333ff]液相色谱[/color][/url]法、[url=https://insevent.instrument.com.cn/t/Mp][color=#3333ff]气相色谱[/color][/url]-质谱法和[url=https://insevent.instrument.com.cn/t/5p][color=#3333ff]液相色谱[/color][/url]-串联质谱法[/color][/font][font=宋体][sup][size=13px][9][/size][/sup][/font][font=宋体][color=black]。其中,[url=https://insevent.instrument.com.cn/t/5p][color=#3333ff]液相色谱[/color][/url]法-串联质谱法因选择性强、灵敏度高、检出限低而成为测定甲硝唑的优势方法[/color][/font][font=宋体][sup][size=13px][10][/size][/sup][/font][font=宋体][color=black]。本文将蜂蜜用乙酸乙酯萃取,提取液浓缩后经 [/color][/font][color=black]MCS [/color][font=宋体][color=black]固相萃取柱快速富集净化样品的前处理方法,减少前处理的操作步骤,同时降低基质干扰,利用超高效[url=https://insevent.instrument.com.cn/t/5p][color=#3333ff]液相色谱[/color][/url]法—串联质谱法测定蜂蜜中甲硝唑的方法,内标法定量,提高了检测效率,适合大批量样品检测。 [/color][/font][color=black]1.材料与方法[/color][size=16px][color=black] [/color][/size][color=black]1.1 仪器与试剂[/color][color=black]Waters Xevo TQ-S三重四极杆质谱仪(美国Waters),配有电喷雾离子源(ESI) Heidolph Multi Reax全能型振荡器(德国海道夫) 氮吹仪(美国Organomation);高速低温离心机(湘仪) 乙腈、甲醇(色谱纯,德国Merck);甲酸(色谱纯,上海麦克林);乙酸乙酯(色谱纯,美国Fisher);氨水(分析纯,天津科密欧);盐酸(优级纯,北京化工厂);MCS固相萃取小柱(天津,艾杰尔):500ml/6ml;甲硝唑标准品、D4-甲硝唑(纯度均大于99.0%)。实验用水为超纯水(电阻率为18.2mΩ.厘米)。[/color][color=black]1.2 样品前处理[/color][color=black]1.2.1 样品提取 称取蜂蜜5g(精确到 0.01 g)于50ml离心管中,加入100μlD4-甲硝唑内标应用液(20.0ng/ml),加水10ml,混合溶解,再加入10mL乙酸乙酯,涡旋1min,震荡提取 10min,1000rpm 离心 2min,吸取上层乙酸乙酯相 5mL 于10mL 试管,50℃氮气吹干后,加入 0.1mL 甲醇溶解,再加入 1.9mL 40mmol/L盐酸溶液,超声溶解 1min,转入 2mL 离心管,12000rpm 离心 2min,上清液待净化。[/color][color=black]1.2.2 样品净化 依次用 5mL 甲醇、5mL 水、5mL 40mmol/L 的盐酸溶液活化平衡MCS 固相萃取柱,然后转移上述上清液至 MCS 柱内,待样品过柱后,用 5mL水淋洗除杂,真空抽干柱内液体后加入 5mL 乙酸乙酯洗脱,再用 5mL 甲醇淋洗除杂,真空抽干后用 5mL 5%氨化甲醇洗脱,收集于 10mL 具塞试管内,得甲硝唑洗脱液;洗脱液在 50℃下用氮气吹干,分别先加入 0.1mL 甲醇超声溶解残留物,再加入0.9mL 10%甲醇/水溶液混匀,过 0.22μm 滤膜后待 [url=https://insevent.instrument.com.cn/t/Yp][color=#3333ff]LC-MS[/color][/url]/MS 分析。[/color][color=black]1.3 仪器条件[/color][color=black]1.3.1 [url=https://insevent.instrument.com.cn/t/5p][color=#3333ff]液相色谱[/color][/url]条件 色谱柱:Waters ACQUITY UPLC BEH C18(100 mm×2.1 mm,1.7 μm),流动相A为0.05% 氨水溶液,B为乙腈,流速为 0.3 mL/min,柱温:40 ℃,进样量 5.0 μl。[/color][color=black]1.3.2 质谱条件 电喷雾离子源:ESI;质谱多重反应监测方式:MRM;正离子模式(ESI+);毛细管电压:0.5 kV;离子源温度150 ℃;脱溶剂气温度400 ℃;脱溶剂气流量800 L/h。其它质谱参数见表1。[/color][align=center][color=black]表1 [/color]甲硝唑的质谱参数与保留时间[/align][table][tr][td][align=center]化合物名称[/align][/td][td][align=center]母离子[/align][/td][td][align=center]子离子[/align][/td][td][align=center]碰撞能量(eV)[/align][/td][td][align=center]锥孔电压(V)[/align][/td][td][align=center]保留时间(min)[/align][/td][/tr][tr][td][align=center]甲硝唑[/align][/td][td][align=center]172.2[/align][/td][td][align=center]128.1*[/align][align=center]82.1[/align][/td][td][align=center]18[/align][align=center]20[/align][/td][td][align=center]54[/align][/td][td][align=center]1.40[/align][/td][/tr][tr][td][align=center]D4-甲硝唑[/align][/td][td][align=center]176.2[/align][/td][td][align=center]128.1*[/align][align=center]49.0[/align][/td][td][align=center]22[/align][align=center]22[/align][/td][td][align=center]2[/align][/td][td][align=center]1.39[/align][/td][/tr][/table]注:*为定量离子[color=black]结果与讨论[/color][color=black] 前处理方法优化 针对蜂蜜样品和目标物的性质,比较了3种不同的前处理方式,包括:(1)采用水直接溶解蜂蜜,再将蜂蜜水溶液进行固相萃取净化;(2)加水溶解蜂蜜后,加入乙酸乙酯萃取目标物,取乙酸乙酯层并将溶剂吹干后加入超纯水溶解残渣,再进行固相萃取净化;(3)采用pH=8.8的磷酸缓冲液溶解蜂蜜,再将样品溶液进行固相萃取净化.通过加标回收实验比较回收率表明,本实验采用方法(2)的回收率明显高于其他2种方式,故对蜂蜜试样采用方法(2)前处理方式。[/color][color=black] 基质效应的影响 基质和干扰组分的存在影响待测物的离子化效率,从而影响定量结果的准确性,常表现为基质增强或基质抑制效应[/color][sup][size=13px][11][/size][/sup][color=black]。分别采用空白蜂蜜,按照实验方法提取与净化后的定容液和初始流动相作为标准溶液的稀释溶剂,通过测定标准溶液的峰面积的比值考察基质效应的强弱。结果表明:两者的峰面积比值为0.757,即蜂蜜基质对甲硝唑的测定具有一定的抑制效应,本实验选择同位素内标法定量,从而有效地降低样品的基质效应的对测定结果的影响。[/color][color=black]2.3 质谱条件的优化 将甲硝唑标准工作液注入质谱,启用质谱智能方法开发程序,优化碰撞能量,碰撞池电压等参数,进一步优化其他质谱参数使灵敏度和离子化效率达到最优时保存为质谱方法。离子对、碰撞能量、锥孔电压、电离方式见表1。[/color][color=black]2.4 方法的线性关系和检出限 以甲硝唑与相应同位素内标的色谱峰面积比(y)为纵坐标,以甲硝唑的质量浓度(x)为横坐标,绘制工作曲线,线性回归方程为Y=1.004X+0.1243,相关系数r:0.9996,线性关系良好。以信噪比S/N=3时对应的浓度为方法检出限为0.05[/color]μg/kg[color=black],S/N=10时对应的浓度为方法定量限为0.15[/color]μg/kg。标准工作曲线见图1。[align=center][color=black]图1 甲硝唑工作曲线[/color][/align][color=black]2.5 方法的精密度和回收率 [/color]以5g空白蜂蜜样品作为本底,分别加入高、中、低3种不同浓度标准应用液,得到浓度为1[color=black]μg/kg[/color]、5μg/kg、20μg/kg的加标样品,充分混匀后按样品处理方法进行处理,平行测定6次,计算其加标回收率和相对标准偏差(RSD),加标回收率分别为87%~96.3%,RSD在2.23~6.17%之间,结果表明,此方法具有良好的准确性和精密度。[color=black]2.6 样品检测[/color][font=calibri][size=13px] [/size][/font][color=black]采用本方法对市售30份不同蜂蜜样品进行检测,其中1份检出甲硝唑残留,含量是0.27 [/color]μg/kg,检出率为3.3%。3 结论本研究建立了超高效[url=https://insevent.instrument.com.cn/t/5p][color=#3333ff]液相色谱[/color][/url]法—串联质谱法测定蜂蜜中甲硝唑的含量的方法,样品前处理采用乙酸乙酯提取,固相萃取柱富集和净化,净化效果好,提取效率高。不同蜂蜜样品基质效应使甲硝唑在质谱中存在不同程度的基质抑制效应,实际测定中蜂蜜的种类繁多,若使用外标法定量应尽量使用与待测样品基质相同的样品作基质匹配工作曲线,基质不同需要配置不同的曲线系列,大大增加了工作量。本研究采用同位素内标法定量,降低了样品的基质效应的影响,只需配置一套工作曲线,提高了工作效率。本方法快速、准确、灵敏,能够满足日常蜂蜜样品中甲硝唑残留的大批量检测。参考文献[1]梁明.超高效[url=https://insevent.instrument.com.cn/t/5p][color=#3333ff]液相色谱[/color][/url]-串联质谱法对蜂蜜中氯霉素和甲硝唑残留的测定分析[J].中国高新科技,2019(17):72-73.[2]张晓艺,张秀尧,蔡欣欣,李瑞芬.超高效[url=https://insevent.instrument.com.cn/t/5p][color=#3333ff]液相色谱[/color][/url]联用三重四极杆质谱法同时测定蜂蜜中氯霉素、甲硝唑和林可霉素[J].预防医学,2019,31(02):212-216.[3]周贻兵,吴坤,李磊,林野,刘利亚.超高效[url=https://insevent.instrument.com.cn/t/5p][color=#3333ff]液相色谱[/color][/url]-串联质谱法测定蜂蜜中甲硝唑[J].理化检验(化学分册),2017,53(08):946-949.[4]丁燕玲,陈彤,黄婷,钟名琴,吴雯娟,罗燕.超高效[url=https://insevent.instrument.com.cn/t/5p][color=#3333ff]液相色谱[/color][/url]-串联质谱法测定鸡肉中甲硝唑、二甲硝唑及其代谢物的方法研究[J].广东化工,2018,45(13):245-248+252.[5]王春民,张秋萍,吴春霞.超高效[url=https://insevent.instrument.com.cn/t/5p][color=#3333ff]液相色谱[/color][/url]-串联质谱法检测蜂蜜中的甲硝唑含量[J].食品安全质量检测学报,2016,7(05):1813-1817.[6]章剑,李昌安,李建伟,董骏,张克才.固相萃取-超高效[url=https://insevent.instrument.com.cn/t/5p][color=#3333ff]液相色谱[/color][/url]-串联质谱法同时测定蜂蜜样品中氯霉素和甲硝唑[J].安徽预防医学杂志,2018,24(01):16-20.[7]刘伟,张楠,李兵,范赛,屠瑞莹,吴国华,薛颖,赵榕.固相萃取-超高效[url=https://insevent.instrument.com.cn/t/5p][color=#3333ff]液相色谱[/color][/url]-同位素稀释串联质谱法测定蜂蜜中的甲硝唑和氯霉素[J].分析科学学报,2017,33(01):145-148.[8]肖国军,蔡超海,王生,覃玲.固相萃取高效[url=https://insevent.instrument.com.cn/t/5p][color=#3333ff]液相色谱[/color][/url]串联质谱法同时测定蜂蜜中甲硝唑、氯霉素、甲砜霉素和氟甲砜霉素残留[J].中国卫生检验杂志,2018,28(01):22-25.[9]高何刚,杜赛,王瑞,陈理.超高效[url=https://insevent.instrument.com.cn/t/5p][color=#3333ff]液相色谱[/color][/url]-串联质谱法测定蜂蜜中氯霉素和甲硝唑残留[J].预防医学,2017,29(09):969-972.[10]高何刚.超高效[url=https://insevent.instrument.com.cn/t/5p][color=#3333ff]液相色谱[/color][/url]一串联质谱法测定蜂蜜中氯霉素和甲硝唑残留[J].广东化工,2017,44(15):255-256.[11]图雅,崔建平,赵宏.同位素内标-超高效[url=https://insevent.instrument.com.cn/t/5p][color=#3333ff]液相色谱[/color][/url]-串联质谱法测定蜂蜜中氯霉素及甲硝唑[J].中国食品卫生杂志,2017,29(04):450-453.
【序号】:1【作者】:周杨; 冯群科; 朱永林;【题名】:高效液相色谱-串联质谱法测定饲料中三聚氰胺【期刊】:中国饲料【年、卷、期、起止页码】:2010年 12期 【全文链接】:http://202.119.208.220:8002/kns50/detail.aspx?dbname=CJFD2010&filename=SLGZ201012013
序号:作者:倪姮佳; 黄显会; 方炳虎; 贺利民; 赵永达;期刊:分析测试学报,题名:高效液相色谱-串联质谱法测定猪组织中的土拉霉素年号:2011年03期链接:http://202.119.208.220:8002/kns50/detail.aspx?dbname=CJFDTEMP&filename=TEST201103019
GB 29704-2013 食品安全国家标准 动物性食品中环丙氨嗪及代谢物三聚氰胺多残留的测定 超高效液相色谱-串联质谱法
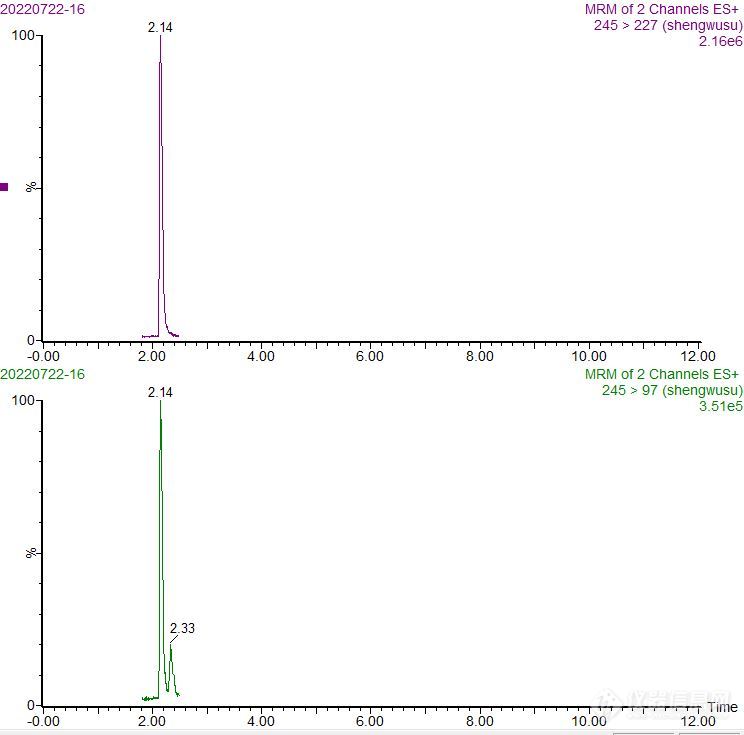
[size=16px]超高效[url=https://insevent.instrument.com.cn/t/5p][color=#3333ff]液相色谱[/color][/url]-串联质谱法测定奶粉中生物素的含量[/size][align=center][size=16px]户江涛[/size][/align][align=center][size=16px](黑龙江省农垦科学院测试化验中心,黑龙江 佳木斯 154007 )[/size][/align][size=16px]摘要:采用超高效[url=https://insevent.instrument.com.cn/t/5p][color=#3333ff]液相色谱[/color][/url]-串联质谱法建立了检测奶粉中生物素含量的分析方法,对试样提取、净化条件,流动相、色谱柱和质谱条件进行了优化,结果表明该方法与国标微生物法对同一样品检测得到的生物素含量基本一致,但检测所需时间大大减少,且抗干扰能力、精密度均比微生物法高,特别适和大批量奶粉中生物素含量检测。关键词:超高效[url=https://insevent.instrument.com.cn/t/5p][color=#3333ff]液相色谱[/color][/url]-串联质谱;奶粉;生物素生物素又称维生素B7,是生物体内羧基转化酶作用的一种辅酶,在人体生长、代谢、发育过程中发挥着重要的作用。人类自身不能合成生物素,需从膳食中获得,而奶粉是人类(特别是婴幼儿)获取生物素的重要途径,准确测定奶粉中生物素含量有重要意义。目前国家标准规定的生物素测定方法《GB 5009.259-2016 食品安全国家标准 食品中生物素的测定》为微生物法。该方法需要购买特定菌种,成本较高,且菌种难保存、易受污染,实验操作复杂、费时费力、技术难度大、对检验人员和实验室要求较高,且容易受到基质干扰、检测结果重复性较差。同时奶粉成分复杂,所含生物素含量极低,一般为十几个微克/100克。因此,制定一种准确、高效、便捷、灵敏度高的生物素测定方法迫在眉睫。基于高效[url=https://insevent.instrument.com.cn/t/5p][color=#3333ff]液相色谱[/color][/url]的分离能力和质谱的高灵敏度、高选择性,采用[url=https://insevent.instrument.com.cn/t/5p][color=#3333ff]液相色谱[/color][/url]串联质谱测定法具有前处理简单、分析速度快,适用的基质范围广、实用性强,可以为奶粉中生物素含量的测定提供一种有效的检测手段。本文建立的超高效[url=https://insevent.instrument.com.cn/t/5p][color=#3333ff]液相色谱[/color][/url]-串联质谱法测定奶粉中生物素含量[color=black]的方法前处理过程简便、分析时间短、灵敏度高、抗干扰能力强,特别适用于大批量奶粉样品中生物素[/color]含量的检测。1 实验部分1.1 材料与试剂[color=black]生物素(纯度[/color][font=宋体][color=black]≥[/color][/font][color=black]99%,Sigma公司);婴儿配方乳粉定量分析质控样品(BQC1051147452,北京普天同创生物科技有限公司);乙腈、甲酸(色谱纯,Fisher公司);Prime HLB固相萃取柱(200 mg,3 mL,[/color][font=宋体]Waters[/font][color=black]公司);0.2 um有机系滤膜;实验用水为Millipore纯水仪制备。[/color]1.2 仪器与设备UPLC XEVO TQ-S超高效[url=https://insevent.instrument.com.cn/t/5p][color=#3333ff]液相色谱[/color][/url]串联质谱仪(Waters公司);涡旋振荡器。1.3 [color=black]生物素[/color]标准储备液的配置称取一定量生物素[color=black]标准品[/color],用50%乙醇-水溶液配置成质量浓度为100 ug/mL标准储备液,于2~4℃冰箱保存(有效期1个月),待用;临用前将溶液回温至室温,并吸取一定体积储备液用水逐级稀释成所需浓度的标准工作液。1.4 样品前处理准确称取1.00 g(精确到0.01 g)奶粉试样于50 mL离心管中,加入10.00 mL纯水涡旋混匀2 min,然后加入10.00 mL乙腈,涡旋混匀1 min,然后在离心机中以15000 r/min离心5 min,取出后吸取2 mL上清液置于[color=black]Prime HLB固相萃取柱中,使其自然流出弃去最初几滴,然后用玻璃试管接取流出液约1 mL涡旋混匀,[/color]过0.22[font=宋体]u[/font]m有机系微孔滤膜后供UP[url=https://insevent.instrument.com.cn/t/Yp][color=#3333ff]LC-MS[/color][/url]/MS分析测定。1.5 [url=https://insevent.instrument.com.cn/t/5p][color=#3333ff]液相色谱[/color][/url]及质谱条件[url=https://insevent.instrument.com.cn/t/5p][color=#3333ff]液相色谱[/color][/url]:色谱柱:Waters HSS [font=times new roman]T3(1.8 μm,100mm×2.1mm);柱温:30℃[/font];流速:[font=times new roman]0.3 [/font]mL/min;进样量:[font=times new roman]2[/font] [font=times new roman]μL;流动相A:乙腈;流动相B:0.1%的甲酸水溶液。梯度洗脱程序:0~0.5min,10% A;0.5~3. 0 min,10%~100% A;3. 0 ~4. 0 min,100%A,4 ~4.1min,100% A~10% A,4.1 ~5.0min 10% A。[/font]质谱:离子源:电喷雾离子源( ESI [sup]+[/sup] ) ;扫描方式:正离子扫描;检测方式:多反应监测( MRM);毛细管电压:3.2 kv;离子源温度:150℃;去溶剂气温度:500℃;去溶剂气流量:1000 L /h;定性、定量离子对及碰撞能量见表1。[/size][align=center][size=16px]表1生物素的质谱参数[/size][/align][table][tr][td][align=center][size=16px]分析物[/size][/align][/td][td][align=center][size=16px]锥孔电压/V[/size][/align][/td][td][align=center][size=16px]母离子/(m/z)[/size][/align][/td][td][align=center][size=16px]子离子/(m/z) [/size][/align][/td][td][align=center][size=16px]碰撞能量/V[/size][/align][/td][/tr][tr][td][size=16px]生物素[/size][/td][td][align=center][size=16px]30[/size][/align][size=16px][/size][/td][td][align=center][size=16px]245[/size][/align][size=16px][/size][/td][td][align=center][size=16px]227﹡[/size][/align][align=center][size=16px]97[/size][/align][size=16px][/size][/td][td][align=center][size=16px]13[/size][/align][align=center][size=16px]25[/size][/align][size=16px][/size][/td][/tr][/table][size=16px]﹡为定量离子2 结果与讨论2.1 色谱质谱条件及前处理过程的优化流动相的选择:对比了酸性体系(0.1%甲酸水溶液)与甲醇、乙腈的流动相体系组合,结果发现生物素在乙腈体系中响应值比甲醇更好一些,故本研究采用0.1%甲酸水溶液+甲醇流动相体系。色谱柱的选择:比较了[font=宋体]Waters [/font]BEH C[sub]18[/sub](1.7 μm,50mm×2.1mm)和[font=宋体]Waters [/font]HSS T[sub]3[/sub](1.8 μm,100mm×2.1mm)两种不同填料的分析柱,实验时发现目标物在这两款色谱柱上响应值差不多,但目标物在BEH C[sub]18[/sub]上保留时间比HSS T[sub]3[/sub]要短,考虑到生物素本身属于水溶性维生素,极性较强,若出峰太早可能造成奶粉中一些极性强的基质随目标物一起共流出进而干扰目标物测定,因此本方法采用了HSS T[sub]3[/sub]色谱柱。质谱参数优化:将1.0 mg/L 生物素标准溶液直接注射到质谱中,在正离子模式下进行母离子全扫描,发现目标物各自对应的准分子离子峰[M+H][sup]+[/sup]具有很好的响应,然后在分别进行子离子全扫描,各得到两对丰度高、干扰小的子离子对进行MRM监测,最终确定的质谱条件见表1,相应的色谱质谱图见图1、图2。前处理过程优化:生物素属于水溶性维生素,用纯水作为提取试剂可以得到很好的提取效果。但实验过程中发现,用纯水将奶粉溶解后整个溶液呈乳白色,只通过离心方式很难去除其中大量的蛋白、脂肪等杂质,需要对提取液进行除蛋白操作。通过考察乙酸铅、三氯乙酸、乙腈等几种常用的沉淀蛋白方法,综合考虑在去除蛋白的同时要尽可能减少其它杂质的引入,因此本方法采用乙腈除蛋白的方式,比较了几种不同水/乙腈比例,最终选定水/乙腈(1:1体积比)达到最优的实验效果。对于脂肪的去除则选用了目前较流行的[color=black]Prime HLB固相萃取柱通过式方法,即提取液通过Prime HLB时脂肪等大分子保留在SPE小柱上,目标物不保留以达到去除脂肪等杂质的目的,[/color]综合以上因素本实验最终采用了1.4的前处理方法。[/size][align=center][size=16px][img]https://ng1.17img.cn/bbsfiles/images/2022/10/202210071506558084_124_1729077_3.jpg[/img][/size][/align][align=center][size=16px]图1 [color=black]生物素[/color]标准溶液(10 ng/mL)MRM色谱图[/size][/align][size=16px][/size][align=center][size=16px][img]https://ng1.17img.cn/bbsfiles/images/2022/10/202210071506561980_1283_1729077_3.jpg[/img][/size][/align][align=center][size=16px]图2 奶粉样品中[color=black]生物素[/color]MRM色谱图[/size][/align][size=16px][color=black]2.2 线性范围和定量限[/color][color=black]吸取不同体积的生物素标准储备液(1.3),用[/color]纯水[color=black]分别配置不同浓度的[/color]上机标准溶液,以各自定量离子的峰面积(或与内标峰面积比值)为Y对应质量浓度X([color=black]m[/color]g/L)做标准曲线,得到的线性方程和相关系数见表2;以10倍信噪比(S/N)计算得到生物素的定量下限,结果见表2。表2 生物素标准溶液的线性方程、相关系数和定量下限(LOQ)[/size][table][tr][td][align=center][size=16px]分析物[/size][/align][/td][td][align=center][size=16px]线性范围/(ng/mL)[/size][/align][/td][td][align=center][size=16px]线性方程[/size][/align][/td][td][align=center][size=16px]R[/size][/align][/td][td][align=center][size=16px]LOQ/(ug/100g)[/size][/align][/td][/tr][tr][td][align=center][size=16px]生物素[sub] [/sub][/size][/align][/td][td][align=center][size=16px]0.2~50[/size][/align][size=16px][/size][/td][td][align=center][size=16px]Y=3078.1X-106.32[/size][/align][size=16px][/size][/td][td][align=center][size=16px]0.9993[/size][/align][size=16px][/size][/td][td][align=center][size=16px]0.5[/size][/align][/td][/tr][/table][size=16px][color=black]2.3回收率和精密度[/color][color=black]生物素在奶粉中天然存在[/color],选取已知生物素含量的奶粉作为基质进行加标。具体添加水平为:[color=black]0.5,5,50[/color] ug/100g。[color=black]每个[/color]水平重复6次,[color=black]同时做该奶粉的本底实验。[/color]按照1.4前处理方法处理后上机检测,回收率计算结果(扣除空白后)见表3。结果表明,该方法生物素的平均回收率为87.2%~110%,相对标准偏差(RSD,n=6)为2.3%~5.2%,均满足实验要求。[/size][align=center][size=16px]表3 奶粉生物素的加标回收率和相对标准偏差(n=6)[/size][/align][table][tr][td][align=center][size=16px]分析物[/size][/align][/td][td][align=center][size=16px]添加水平(ug/100g)[/size][/align][/td][td][align=center][size=16px]回收率/%[/size][/align][/td][td][align=center][size=16px]相对标准偏差/%[/size][/align][/td][/tr][tr][td][align=center][size=16px]生物素[/size][/align][size=16px][sub] [/sub][/size][/td][td][align=center][size=16px]0.5[/size][/align][align=center][size=16px]5[/size][/align][align=center][size=16px]50[/size][/align][/td][td][align=center][size=16px]86.8[/size][/align][align=center][size=16px]93.2[/size][/align][align=center][size=16px]91.6[/size][/align][size=16px][/size][/td][td][align=center][size=16px]4.6[/size][/align][align=center][size=16px]3.3[/size][/align][align=center][size=16px]2.1[/size][/align][size=16px][/size][/td][/tr][/table][size=16px][color=black]2.4实际样品分析[/color][color=black]为进一步验证该方法的准确性,采用本方法和《[/color]GB 5009.259-2016[color=black]》微生物法同时对北京普天同创生物科技有限公司的奶粉质控样品BQC1051147452生物素含量进行检测,结果见表4[/color]。由表4可知,UP[url=https://insevent.instrument.com.cn/t/Yp][color=#3333ff]LC-MS[/color][/url]/MS法测定结果与国标方法的结果基本一致,无显著性差异,但前者所需时间更短,精密度更好。[/size][align=center][size=16px]表4 奶粉质控样品[color=black]BQC1051147452[/color]生物素的测定结果[/size][/align][table][tr][td][align=center][size=16px]检测方法[/size][/align][/td][td][align=center][size=16px]特性值区间(ug/100g)[/size][/align][/td][td][align=center][size=16px]测定平均值(n=6)[/size][/align][/td][td][align=center][size=16px]相对标准偏差/%(n=6)[/size][/align][/td][/tr][tr][td][size=16px]UP[url=https://insevent.instrument.com.cn/t/Yp][color=#3333ff]LC-MS[/color][/url]/MS法微生物法[sub] [/sub][/size][/td][td][align=center][size=16px]15.6~22.4[/size][/align][align=center][size=16px]15.6~22.4[/size][/align][size=16px][/size][/td][td][size=16px]18.718.1[/size][/td][td][align=center][size=16px]2.5[/size][/align][size=16px] 4.6[/size][/td][/tr][/table][size=16px]3 结语本文建立了超高效[url=https://insevent.instrument.com.cn/t/5p][color=#3333ff]液相色谱[/color][/url]-串联质谱法(UP[url=https://insevent.instrument.com.cn/t/Yp][color=#3333ff]LC-MS[/color][/url]/MS)测定奶粉中[color=black]生物素[/color]含量的分析方法。该方法具有较高的灵敏度、准确度和精密度,前处理步骤简单,分析速度快,特别适合大批量样品的检测。参考文献:[1] GB 5009.259-2016 食品安全国家标准 食品中生物素的测定.[2] 薛霞, 赵慧男, 魏莉莉, 等. 超高效[url=https://insevent.instrument.com.cn/t/5p][color=#3333ff]液相色谱[/color][/url]-串联质谱法测定蜂蜜中五种水溶性维生素的含量[J]. 食品与发酵工业. 2021,47(12) : 250-256.[3] 李佳兴, 周利, 金艳, 等. 超高效[url=https://insevent.instrument.com.cn/t/5p][color=#3333ff]液相色谱[/color][/url]-串联质谱法测定枸杞子中8种水溶性维生素[J]. 食品科技. 2018,43(11) : 336-341.[/size]
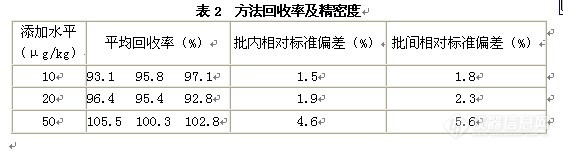
高效液相色谱串联质谱测定猪肉中新型兽药泰拉霉素摘要 建立了猪肉中新型兽药泰拉霉素的液相色谱串联质谱(HPLC-MS/MS)联用确证方法。样品用甲醇+0.1%磷酸(70:30 v/v)提取,离心后用PCX固相萃取小柱净化,Symmetry®C8色谱柱分离,串联质谱多反应监测(MRM)模式下分析,内标法定量。方法的线性范围为10~500μg/kg,检出限为5.0μg/kg,在10、20和50μg/kg 3个浓度水平进行添加实验,平均回收率为93.1%~105.5%,批内相对标准偏差为1.5%~4.6%,批间相对标准偏差为1.8 %~5.6%。 关键词 高效液相色谱串联质谱,猪肉,泰拉霉素,残留 泰拉霉素(Tulathromycin)是一种新近上市且为动物专用的大环内酯类半合成抗生素,分子式为C41H79N3O12(分子量806)。我国农业部在2008年第957号公告中首次允许泰拉霉素在动物生产中使用。泰拉霉素主要用于放线杆菌、支原体、巴氏杆菌、副嗜血杆菌引起的猪、牛的呼吸系统疾病。具有用量少、一次给药、低残留和动物专用等众多优点。在我国,大环内酯类药物现行使用较为广泛的是泰乐菌素和替米考星,虽然这2种药物在生产中都取得了良好的效果,但随着使用时间的延长,我国很多地区出现了不同程度的耐药性,导致用量不断增大,但治疗效果却在逐步降低 ,而泰拉霉素药效均强于泰乐菌素和替米考星等市场广泛使用的大环内酯类药物。因此,泰拉霉素在畜禽生产中使用前景非常广阔,其残留分析方法研究也显得尤为必要。 目前国内外大环内酯类药物残留检测方法已有ELISA筛选法、薄层色谱法、气质联用法、高效液相色谱和高效液相色谱串联质谱法等众多方法。但泰拉霉素的残留检测方面研究较少,国内尚未见有相关报道。本研究建立了以罗红霉素(C41H76N2O15, 837)为内标的泰拉霉素HPLC-MS/MS方法,方法的定量限为10μg/kg,可以满足国内外有关法规对其残留检测的要求,可为残留监控提供技术支持。1 材料与方法1.1仪器与材料 Waters 2695 Quattro MicroTM API高效液相色谱串联质谱仪(美国Waters公司);固相萃取仪(美国SUPELCO公司)。 泰拉霉素标准品(Sigma公司);罗红霉素 (Sigma公司);乙腈(色谱纯);甲醇(色谱纯);其余试剂均为分析纯试剂。PCX固相萃取柱(3 mL, 60 mg,AGELA公司)。1.2标准溶液的配制 泰拉霉素和内标储备液:分别准确称取10mg标准品于10mL容量瓶中,用0.05M K2HPO4/乙腈(75:25,v/v; pH6)定容,混匀后置冰箱冷藏储存,有效期3个月。 标准工作溶液的配制:吸取储备液1mL于100mL容量瓶中并用0.05M K2HPO4/乙腈(75:25,v/v; pH6)定容,再从中吸取1mL于50mL容量瓶中得0.2mg/L,用于添加(置冰箱冷藏储存,有效期2个月)。内标物罗红霉素的添加溶液配制方法同泰拉霉素。1.3样品制备 称取2.0g组织样品于50mL的聚四氟乙烯塑料管中,加入10mL甲醇+0.1%磷酸提取液(70:30 v/v)和50μL内标工作液,匀质1 min后5000rpm转速离心2min,收集上清液过已经分别用3mL甲醇和3mL提取液润洗过的60mg/3mL的PCX小柱,待全部过柱后,再用3mL水、3mL甲醇淋洗,最后用4mL4%的氨化甲醇洗脱,50℃水浴氮气吹干后用1mL流动相定容,进行HPLC-MS/MS分析。1.4仪器分析条件 液相色谱条件:Symmetry®C8 5μm 3.9mmx20mm,流动相:乙腈:0.1%甲酸水溶液=70:30;柱温:30℃,进样室温度15℃,进样量:10μL,流速为0.3mL/min。质谱条件:电离模式:ESI(+);检测方式:多级反应检测(MRM);毛细管电压:4.2 KV;锥孔电压:20 V;RF透镜电压:0.2 V;离子源温度:110 ℃;脱溶剂气温度:350 ℃;锥孔气流速:100 L/h;脱溶剂气流速:600 L/h;二级碰撞气:氩气。1.5添加回收率实验 添加回收率实验添加浓度为10、20和50µg/kg,实验步骤同1.3。2 结果与讨论2.1样品的提取和净化 泰拉霉素含3个氨基基团,为弱碱性化合物,易溶于酸性溶液和极性溶剂中,在pH 6~8的水溶液中较稳定,在pH 9条件下均不稳定。对泰拉霉素残留的提取和净化方法的设计主要依据其弱碱性、脂溶性和酸碱不稳定性。本实验比较了4种提取液:乙腈-0.1%偏磷酸(70:30 v/v)、甲醇-0.1%偏磷酸(70:30 v/v)、乙腈-0.1%磷酸(70:30 v/v)、甲醇-0.1%磷酸(70:30 v/v)对泰拉霉素的提取效果。结果表明,甲醇-0.1%磷酸(70:30)提取液对泰拉霉素的提取效果较好,回收率高于其他提取液,故本方法选择甲醇-0.1%磷酸(70:30)为样品提取液。 液-液分配(LLE)和固相萃取(SPE)是大环内酯类药物的2种主要净化手段,其中LLE操作较麻烦且回收率较低、重复性较差,所以本研究采用固相萃取技术净化,并比较了3种SPE净化小柱:SUPELCO C18小柱、Oasis MCX小柱和PCX小柱, 由于Oasis MCX小柱和PCX小柱兼有阳离子交换和疏水作用机制,基于泰拉霉素的弱碱性和脂溶性的理化特性,Oasis MCX小柱和PCX小柱的净化效果好于C18小柱。Oasis MCX小柱和PCX小柱提取效率没有明显差别,但PCX小柱成本低,所以,本方法净化选择PCX小柱。2.2色谱质谱条件的优化[fo
近日,中国国家标准化管理委员会(以下简称“国标委”)发布了《液相色谱-串联四极质谱仪性能的测定方法》(GB/T 35410-2017)。该国家标准收录在2017年第32号中国国家标准公告中,将于2018年4月1日开始实施。该标准由国家科技部提出,由全国仪器分析测试标准化技术委员会归口,起草单位是中国计量科学研究院。该国家标准规定了液相色谱-串联四极质谱仪性能的测试方法,适用于液相色谱-串联四极质谱仪性能的测定。 液相色谱-串联四极质谱仪作为最有代表性的[url=https://insevent.instrument.com.cn/t/Yp][color=#3333ff]液质联用[/color][/url]类型,广泛应用于食品、药品、环境、化工、临床、科研等领域,几乎覆盖了国计民生的方方面面。2017年,我国采购的液相色谱-串联四极质谱仪总量超过1000台,总金额约在15亿元到20亿元之间。目前,我国尚不具备成熟的[url=https://insevent.instrument.com.cn/t/Yp][color=#3333ff]液质联用仪[/color][/url]生产能力,主要靠进口。目前市场上液相色谱-串联四极质谱仪的主流品牌多达6-7家,型号更是繁多,普通购买者没有办法快速、直观地了解每台仪器的性能。该国家标准的出台,树立了统一的仪器性能评价标准,有助于对不同品牌、型号的仪器参比和行业秩序的改善。也有助于产品研发时做技术评价。 以下为详细内容:[url]http://www.instrument.com.cn/news/20180126/238974.shtml[/url]
请问各位老师,Waters[url=https://insevent.instrument.com.cn/t/5p][color=#3333ff]液相色谱[/color][/url]串联质谱仪有Xevo TQ-XS、Xevo TQ-S、Xevo TQ-S micro等几个型号,求教这三个型号的差异,谢谢
GBT30926-2014化妆品中7种维生素C衍生物的测定 高效液相色谱-串联质谱法
[img]http://www.instrument.com.cn/bbs/images/affix.gif[/img][url=http://www.instrument.com.cn/bbs/download.asp?ID=102920]动物组织中盐酸吗啉胍残留的高效液相色谱-串联质谱测定[/url]
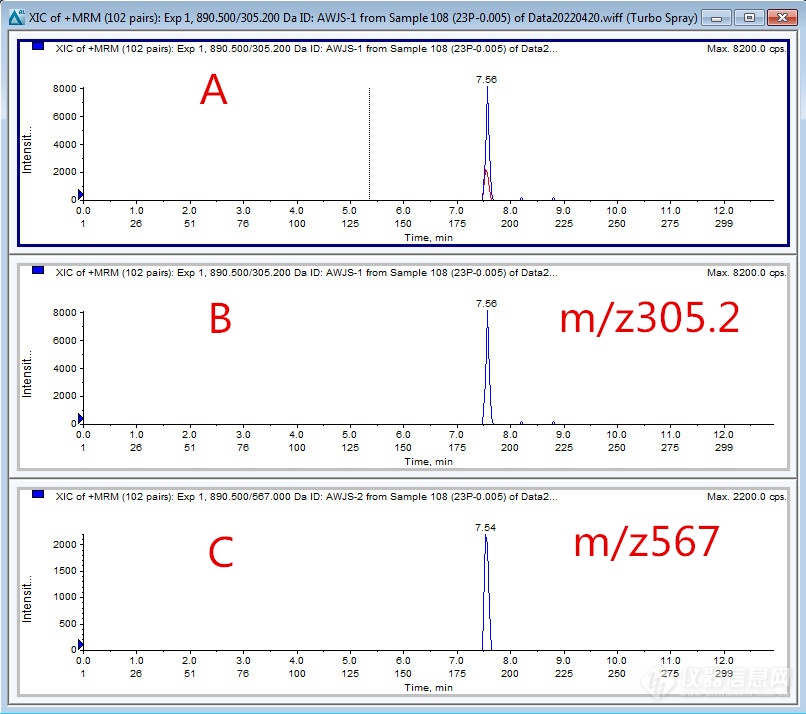
[align=center][size=24px]超高效[url=https://insevent.instrument.com.cn/t/5p][color=#3333ff]液相色谱[/color][/url]-串联质谱检测农产品中阿维菌素残留方法研究[/size][/align][align=center][size=18px]鹤壁市农产品检验检测中心 张艳丽[font=宋体] 王丽娟[/font][/size][/align][align=left] 阿维菌素是一种新型类广谱性杀虫杀螨剂,由阿维链霉素经液体发酵加工而成,具有高效、广谱、有效期长、不易产生抗药性等特点,已经作为高毒有机磷农药的替代品而广泛应用。目前阿维菌素已广泛用于要水果样品中果树(苹果、梨、桃)、蔬菜(黄瓜、番茄)有害生物的防治,GB 2763-2021《食品安全国家标准 食品中农药最大残留限量》中阿维菌素的残留限量(MRL)要求很高,规定其在柑桔、梨和黄瓜中的MRL为0.02mg/kg,叶菜和豆中为0.05mg/kg。目前阿维菌素的检测方法主要有高效液相色谱法(HPLC)[sup][1-5][/sup]、酶联免疫法(ELISA)、液质联用仪法(LC-MS)[sup][6-10][/sup]等。近几年液相色谱质谱法应用日益广泛,它可以提高目标物质的灵敏度以及回收率,缩短进样时间,提高检测效率。但在实际工作中,阿维菌素的响应值低、灵敏度低、标准曲线线性差等问题一直存在,本文应用超高效液相色谱-串联质谱仪(UPLC-MS/MS),对阿维菌素的检测条件进行色谱、质谱条件的优化,可以在检测农产品中阿维菌素时,得到快速、准确的检测方法。[/align]1 实验部分1.1 仪器与试剂AB Sciex4500高效液相色谱-串联质谱仪(配有电喷雾电离(ESI)源,美国AB SCIEX公司);GL-21M高速冷冻离心机(湖南湘仪);混匀器(德国heidolph公司);涡旋混匀器(德国IKA公司)。阿维菌素(100ug/mL)甲醇作为溶剂,购于农业部环境保护科研监测所;乙腈(HPLC级,上海安谱公司);甲醇(HPLC级,美国Merck公司)。甲酸(LCMS,美国Fisher公司);甲酸铵(LCMS,美国Fisher公司);十八烷基键合硅胶(C18)、N-丙基乙二胺(PSA)、无水MgSO[sub]4 [/sub][font=calibri]、[/font]NaCL均为分析纯(深圳逗点生物)。1.2 标准溶液的配置精确称取一定量的阿维菌素标液,用甲醇溶解后定容,配制成2.0ug/mL贮备溶液,于-20℃避光储存。1.3前处理方法称取10g(精确至0.01g)试样于50mL塑料离心管中,加人10mL乙腈及1颗陶瓷均质子,剧烈振荡1min.加人4g无水硫酸镁、1g氯化钠、1g柠檬酸钠二水合物、0.5g柠檬酸二钠盐倍半水合物,剧烈振荡1min后4200r/min离心5min。吸取8mL上清液至内含除水剂和净化材料的塑料离心管中 对于颜色较深的试样,离心管中另加人GCB,涡旋混匀1min。4200r/min离心5min,吸取上清液过0.22μm有机微孔滤膜后上机测定。1.4 仪器条件条件1.4.1色谱条件色谱柱:Altantis T3柱(150mm×2.1mm,3.0μm);柱温:40℃;流动相:A相为水(含0.01%甲酸(v/v)和1mM/L甲酸铵),B相为甲醇(含0.005%甲酸(v/v)和2mM/L甲酸铵)。柱流速为0.40mL/min。梯度洗脱程序:0~2.0min,90%A;5.0~12min,5%A;12.1~13min,90%A;流速:0.4mL/min;进样量:1μL。1.4.2质谱条件离子源:ESI;扫描方式:正离子扫描;扫描方式:多反应监测(MRM)模式;电喷雾电压:4500V;雾化气压力:50psi;气帘气压力:30psi;辅助加热气:60psi;离子源温度:300℃;碰撞气压力:9psi。2 结果与讨论2.1样品前处理的优化本实验采用GB23200.121《食品安全国家标准植物源性食品中331种农药及其代谢物残留量的测定 液相色谱-质谱联用法》,以乙腈作为提取溶剂,将目标物质、色素等有机物质提出,然后采用饱和盐进行层析分层,用吸附剂进行净化。2.2色谱条件的优化2.2.1色谱柱选择分别用Shimadzu C18(75mmx2.0m,1.6μm)、Agilent EC-C18(100mmx3.0m,2.7μm)、Altlantis T3(150mm×2.1mm,3μm)等3种液相色谱柱对阿维菌素进行响应值与分离效果的比对,所有色谱柱均能出峰,但采用Altlantis T3和Shimadzu C18色谱柱时峰形对称性好,半峰宽窄、且出峰时间相同,但采用Altlantis T3色谱柱时峰面积明显高于Shimadzu C18色谱柱。因此最终选择Altlantis T3色谱柱进行分离。2.2.2流动相选择流动相条件是影响目标化合物的色谱分离和仪器响应的一个重要方面,根据阿维菌素的性质,比较了甲醇和乙腈两种有机相,结果表明,甲醇是质子性溶剂,更易离子化,[M+NH[sub]4[/sub]][sup]+[/sup]峰的响应值要高于乙腈流动相,所以在本实验中选用甲醇和水作为流动相。在水相中加入甲酸铵和甲酸等试剂,是改善色谱峰形、提高仪器响应值和离子化率的常用手段,通常采用酸性流动相有利于在正离子模式下进行质谱检测,试验考察了不同浓度的甲酸与甲酸铵溶液与甲醇组合,发现随着甲酸铵浓度含量增大,阿维菌素的响应值也在增强,若甲酸铵浓度超过2mmol/L时,响应值开始降低。因此,最终选择0.005%甲酸加2mmol/L甲酸铵作为水相。优化后的液相条件下,可得到标准图谱如图1。[align=center][img=,690,611]https://ng1.17img.cn/bbsfiles/images/2022/09/202209011806214247_4665_1645480_3.png!w690x611.jpg[/img][/align] 图1 阿维菌素标液的MRM总离子及选择离子的离子流色谱图(10μg/L)2.3质谱条件的选择2.3.1检测离子的选择阿维菌素的分子式是C[sub]48[/sub]H[sub]72[/sub]O[sub]14[/sub],理论分子量为872.4921。采用母离子扫描(MS Scan),获得一级质谱图,通过分子质量确定阿维菌素多以加合离子[M+NH[sub]4[/sub]][sup]+[/sup]、[M+Na][sup]+[/sup]、[M+H][sup]+[/sup]形式存在,本实验选择离子丰度极强的[M+NH[sub]4[/sub]][sup]+[/sup](m/z890.5)作为母离子。然后,优化毛细管电压等参数,使母离子强度达到最高。选择母离子后,进行子离子扫描(Daughter Scan),获得二级质谱图,得到305.2、567和145.1。进行MRM多反应监测扫描,再次优化碰撞能量,使其离子化效率达到最佳。最终,本实验选择丰度最强、受干扰小的890.5/567作为定性离子对,而890.5/305.2作为定量离子对。阿维菌素检测的质谱参数见表1。 表1 阿维菌素检测的质谱条件[table][tr][td][align=center][color=black]化合物[/color][/align][/td][td][align=center][color=black]母离子/[/color][/align][align=center][color=black](m/z)[/color][/align][/td][td][align=center][color=black]保留时间/min[/color][/align][/td][td][align=center][color=black]产物离子/(m/z)[/color][/align][/td][td][align=center][color=black]碰撞能量/eV[/color][/align][/td][td][align=center][color=black]去簇电压/V[/color][/align][/td][/tr][tr][td][align=center][color=black]阿维菌素[/color][/align][/td][td][align=center][color=black]890.5[/color][/align][/td][td][align=center][color=black]7.56[/color][/align][/td][td][align=center][color=black]305.2[/color][/align][/td][td][align=center][color=black]32[/color][/align][/td][td][align=center][color=black]65[/color][/align][/td][/tr][tr][td] [/td][td] [/td][td] [/td][td][align=center][color=black]567[/color][/align][/td][td][align=center][color=black]19[/color][/align][/td][td][align=center][color=black]65[/color][/align][/td][/tr][/table]2.3.2离子源温度的选择考察了电喷雾离子源(ESI[sup]+[/sup])对阿维菌素的灵敏度影响,结果表明:ESI[sup]+[/sup]源受离子源温度影响比较明显,阿维菌素用的[M+NH4][sup]+[/sup]峰作母离子,温度过高或过低都会抑制目标物离子化,分别用300℃、350℃、400℃离子源温度作了试验,由表2可知:随着离子源温度的升高,响应值越低,当离子源温度为300℃时,灵敏度最高。表2 不同离子源温度的灵敏度[table][tr][td][align=center][color=black]化合物[/color][/align][/td][td][align=center][color=black]母离子/[/color][/align][align=center][color=black](m/z)[/color][/align][/td][td][align=center][color=black]保留时间/min[/color][/align][/td][td][align=center][color=black]产物离子/(m/z)[/color][/align][/td][td][align=center][color=black]离子源温度/℃[/color][/align][/td][td][align=center][color=black]响应强度/%[/color][/align][/td][/tr][tr][td][align=center][color=black]阿维菌素[/color][/align][/td][td][align=center][color=black]890.5[/color][/align][/td][td][align=center][color=black]7.56[/color][/align][/td][td][align=center][color=black]305.2[/color][/align][/td][td][align=center][color=black]300[/color][/align][/td][td][align=center][color=black]3.1e[/color][sup][color=black]4[/color][/sup][/align][/td][/tr][tr][td] [/td][td] [/td][td] [/td][td][align=center][color=black]567[/color][/align][/td][td] [/td][td][align=center][color=black]1.1e[/color][sup][color=black]4[/color][/sup][/align][/td][/tr][tr][td][align=center][color=black]阿维菌素[/color][/align][/td][td][align=center][color=black]890.5[/color][/align][/td][td][align=center][color=black]7.56[/color][/align][/td][td][align=center][color=black]305.2[/color][/align][/td][td][align=center][color=black]350[/color][/align][/td][td][align=center][color=black]2.1e[/color][sup][color=black]4[/color][/sup][/align][/td][/tr][tr][td] [/td][td] [/td][td] [/td][td][align=center][color=black]567[/color][/align][/td][td] [/td][td][align=center][color=black]6000[/color][/align][/td][/tr][tr][td][align=center][color=black]阿维菌素[/color][/align][/td][td][align=center][color=black]890.5[/color][/align][/td][td][align=center][color=black]7.56[/color][/align][/td][td][align=center][color=black]305.2[/color][/align][/td][td][align=center][color=black]400[/color][/align][/td][td][align=center][color=black]2.0e[/color][sup][color=black]4[/color][/sup][/align][/td][/tr][tr][td] [/td][td] [/td][td] [/td][td][align=center][color=black]567[/color][/align][/td][td] [/td][td][align=center][color=black]5000[/color][/align][/td][/tr][/table]2.4标准曲线、线性范围及检出限分别用甲醇配制含量分别为0.005mg/L、0.01mg/L、0.05mg/L、0.1mg/L、0.2mg/L的阿维菌素标准溶液,在上述实验条件下进样1.0μL,以峰面积为纵坐标,浓度为横坐标绘制线性关系曲线,结果表明阿维菌素标准溶液与相对应峰面积呈现良好的线性关系,其线性回归方程:A=6.82989e6+11635.39890,R=0.99913。按上述样品前处理方法及液相色谱检测条件分析得出阿维菌素在农产品样品中的定量限为0.01mg/kg。2.5方法准确度及精密度选取不同的果蔬、食用菌等农产品,进行添加水平为0.01mg/kg、0.05mg/kg、0.10mg/kg等加标回收试验,添加回收率为80%[font=宋体]~[/font]102.5%,RSD1.5[font=宋体]~[/font]8.5%,见表3,其结果满足农药残留检测回收率和相对标准偏差的分析要求。表3 不同农产品样品中阿维菌素添加回收测定结果(n=3)[table][tr][td=1,2][align=center][color=black]样品名称[/color][/align][/td][td=2,1][align=center][color=black]0.01mg/kg[/color][/align][/td][td=2,1][align=center][color=black]0.05mg/kg[/color][/align][/td][td=2,1][align=center][color=black]0.10mg/kg[/color][/align][/td][/tr][tr][td][color=black]回收率/(%)[/color][/td][td][align=center][color=black]RSD/(%)[/color][/align][/td][td][align=center][color=black]回收率/(%)[/color][/align][/td][td][align=center][color=black]RSD/(%)[/color][/align][/td][td][color=black]回收率/(%)[/color][/td][td][align=center][color=black]RSD/(%)[/color][/align][/td][/tr][tr][td][align=center][color=black]西葫芦[/color][/align][/td][td][align=center][color=black]85.6[/color][/align][/td][td][align=center][color=black]5.1[/color][/align][/td][td][align=center][color=black]89.5[/color][/align][/td][td][align=center][color=black]6.0[/color][/align][/td][td][align=center][color=black]91.4[/color][/align][/td][td][align=center][color=black]1.5[/color][/align][/td][/tr][tr][td][align=center][color=black]西红柿[/color][/align][/td][td][align=center][color=black]92.5[/color][/align][/td][td][align=center][color=black]3.5[/color][/align][/td][td][align=center][color=black]94.6[/color][/align][/td][td][align=center][color=black]2.2[/color][/align][/td][td][align=center][color=black]98.1[/color][/align][/td][td][align=center][color=black]3.4[/color][/align][/td][/tr][tr][td][align=center][color=black]芹菜[/color][/align][/td][td][align=center][color=black]90.1[/color][/align][/td][td][align=center][color=black]4.3[/color][/align][/td][td][align=center][color=black]92.1[/color][/align][/td][td][align=center][color=black]5.6[/color][/align][/td][td][align=center][color=black]102.5[/color][/align][/td][td][align=center][color=black]7.8[/color][/align][/td][/tr][tr][td][align=center][color=black]桔子[/color][/align][/td][td][align=center][color=black]86.1[/color][/align][/td][td][align=center][color=black]6.5[/color][/align][/td][td][align=center][color=black]90.3[/color][/align][/td][td][align=center][color=black]7.1[/color][/align][/td][td][align=center][color=black]92.9[/color][/align][/td][td][align=center][color=black]5.9[/color][/align][/td][/tr][tr][td][align=center][color=black]苹果[/color][/align][/td][td][align=center][color=black]82.3[/color][/align][/td][td][align=center][color=black]5.8[/color][/align][/td][td][align=center][color=black]87.8[/color][/align][/td][td][align=center][color=black]4.6[/color][/align][/td][td][align=center][color=black]90.1[/color][/align][/td][td][align=center][color=black]2.2[/color][/align][/td][/tr][tr][td][align=center][color=black]葡萄[/color][/align][/td][td][align=center][color=black]95.4[/color][/align][/td][td][align=center][color=black]3.6[/color][/align][/td][td][align=center][color=black]95.8[/color][/align][/td][td][align=center][color=black]2.0[/color][/align][/td][td][align=center][color=black]91.8[/color][/align][/td][td][align=center][color=black]7.9[/color][/align][/td][/tr][/table]3结论本研究对液相质谱法检测农产品中阿维菌素残留的仪器条件,进行了优化,解决了阿维菌素检出限高、灵敏度低、标准曲线线性差等问题,优化后的仪器条件方法检出限符合标准要求,阿维菌素的灵敏度高,在浓度0.005mg/L[font=宋体]~[/font]0.20mg/L范围内有良好的线性关系;添加回收率和相对标准偏差均符合分析的要求,是比较理想的阿维菌素残留量的分析方法。参考文献:[1]李晶,董丰收,刘新刚.高效[url=https://insevent.instrument.com.cn/t/5p][color=#3333ff]液相色谱[/color][/url]检测梨中阿维菌素残留方法研究[J]. 农药科学与管理,2008,29(2):17-22.[2]谢显传,张少华,王冬生等.柱前行生高效[url=https://insevent.instrument.com.cn/t/5p][color=#3333ff]液相色谱[/color][/url]法测定果蔬产品阿维菌素及其有毒代谢物的残留量[J].中国农业科学,2005,38(11):2254-2260.[3]梁振益,李嘉诚,罗盛旭,等.高效[url=https://insevent.instrument.com.cn/t/5p][color=#3333ff]液相色谱[/color][/url]法检测水果中阿维菌素残留量[J].现代农药,2005,4(4):20-22.[4]张儒令,安凤颖,胡德禹,等.高效[url=https://insevent.instrument.com.cn/t/5p][color=#3333ff]液相色谱[/color][/url]法检测菜豆中阿维菌素残留量[J].现代农业科技,2020(06):106-108.[5]刘桂伶,李婷婷.高效[url=https://insevent.instrument.com.cn/t/5p][color=#3333ff]液相色谱[/color][/url]法检测8种果蔬中阿维菌素残留量的分析方法[J].新疆农业科技,2020(01):38-39.[6]李增梅,邓立刚,赵涉及.超高效[url=https://insevent.instrument.com.cn/t/5p][color=#3333ff]液相色谱[/color][/url]串联质谱法测定苹果和土壤中阿维菌素的残留量[J].分析化学,2010,(10):1505-1509.[7]林涛,邵金良,刘兴勇,等.QuEChERS-超高效[url=https://insevent.instrument.com.cn/t/5p][color=#3333ff]液相色谱[/color][/url]-串联质谱测定蔬菜中41种农药残留[J].色谱,2015,33(3):235-241.[8]王连珠,黄小燕,陈游,等.QuEChERS前处理-[url=https://insevent.instrument.com.cn/t/5p][color=#3333ff]液相色谱[/color][/url]-串联质谱测定果蔬中18种弱酸性农药残留[J].分析测试学报,2014,33(10):1102-1108.[9]李欣,孙素群,张卫锋,等.QuEChERS-[url=https://insevent.instrument.com.cn/t/5p][color=#3333ff]液相色谱[/color][/url]-串联质谱测定蔬菜中56种农药残留[J].现代食品科技,2017,33(10):245-253,177.[10]李瑞雪,王晶蕾,龚慧.超高效[url=https://insevent.instrument.com.cn/t/5p][color=#3333ff]液相色谱[/color][/url]-串联质谱测定蔬菜水果中阿维菌素残留量[J].现代食品,2020,(22):180-182,189.

超高效液相色谱—串联质谱法测定中药材山药中30种农药残留 张彩凤 (浙江捷岛检测科技有限公司,浙江 杭州 310030) 摘要:建立了中药材山药中30种农药的超高效液相-串联质谱检测方法。样品中的农药残留化合物经乙腈提取,BRP固相萃取柱净化,采用 A:缓冲液(0.1 %甲酸溶液含5mmol/L甲酸铵),B:乙腈-缓冲液(95:5)作为流动相进行梯度洗脱,多反应监测模式(MRM),分段时间窗口采集。30种禁用农药在2.5μg▪L-1~75μg▪L-1的线性范围内,线性良好,相关系数在0.9974~1.0000之间。在0.06 mg▪kg-1、0.10 mg▪kg-1、0.12 mg▪kg- 1 3个添加浓度水平进行了回收率试验,平均回收率61 %~108 %,相对标准偏差在0.24%~8.11%(n=6)之间,各农药成分的检出限在0.11μg ▪ kg-1~5μg ▪ kg-1之间。该方法操作简单,灵敏度、准确度和精密度均符合多农残检测技术的要求,可为中药材中农残风险评估研究提供准确有效的数据支持。关键词:超高效液相色谱—串联质谱;山药;农药残留Detection of 30 pesticides residues in traditional Chinesemedicine Dioscorea opposita by ultra performance liquid chromatography-tandemmass spectrometric Zhang Caifeng (Zhejiang Jiedao detection technology co., Ltd,Hangzhou city, Zhejiang Province, China 310030) Abstract: An ultra performance liquidchromatography-tandem mass spectrometric method was developed for thedetermination of thirty pesticides in traditional Chinesemedicine Dioscorea opposita; The pesticides were extracted withacetonitrile,cleaned up by BRP solid phase extraction column; Gradient elutionwas carried out using A:Buffer (0.1% formic acid solution containing 5 mmol/Lammonium formate) and B: acetonitrile - Buffer (95:5) as mobile phases, andMultiple reaction monitoring(MRM) model was used, The data of the detector wascollected by time windows. The recoveries at 3 spiked levels 0.06 mg▪kg-1、0.10mg▪kg-1 and 0.12 mg▪kg- 1 of 30 target compounds were ranged from61% to 108%, with the relative standard debiations(RSD) from 0.11% to 8.11%(n=6), and the detection limit were ranged from 0.11μg ▪kg-1 to 5.0μg ▪kg-1 (S/N=3). This simple ,sensitive and accurate method can be applied to supplying reliable data for thestudy of the risk assessment of traditional Chinese medicine.Key words: ultraperformance liquid chromatography-tandem mass spectrometric ; Dioscorea opposita ; pesticide residues山药(Dioscorea opposita),别名怀山药、淮山药、土薯、山薯、玉延等,主要产于中国华北、西北及长江流域的江西、湖南等地。山药味甘性平,归脾、肺、肾经,具有补脾养胃、生津益肺、补肾涩精等功效,现代药理学研究表明,其具有抗氧化、抗衰老、调节免疫、抗肿瘤、降血糖等作用【1】。为患者或体弱者服用,随着人们对药品安全性要求不断提高,农药残留问题成为人们关注的焦点,由于某些农药具有残留周期长、污染范围广等特点,为保护消费者的健康,建立高灵敏度、高选择性的多农残检测方法,是农药残留检测技术的发展趋势【2】。对“绿色中药材”的生产具有重要意义【3】。农残检测的方法有:薄层色谱法【4】、气相色谱法【5-7】、液相色谱法【8-9】,气质联用法【10-13】、液质联用法【13-18】、免疫分析法【18-19】等。本文通过色谱条件的优化选择,建立了山药中30种农药残留的超高效液相色谱-串联质谱检测法。该方法操作简单、经济实用,灵敏度、准确度和精密度均符合多农残检测技术的要求,可为中药材中农残风险评估研究提供准确有效的数据支持。 1 实验部分1.1 主要仪器和设备超高效液相色谱仪:Acquity UHPLC,含四元梯度泵、样品管理器、柱温箱 ,美国沃特世公司;串联质谱:API 400
[b][color=#444444]实验室准备购买一台高效液相用来检测食品中添加剂和兽药残留等项目,但是在标准中看到添加剂的检测用高效液相色谱仪(带紫外检测器),在药物残留上用的是“液相色谱串联四极杆质谱仪”和高效液相色谱仪。听说现在现在都用质谱联用仪了,请问高效液相色谱仪和质谱联用仪是一回事吗?到底买哪个能够满足要求?[/color][/b]
高效液相色谱串联质谱走标准品,不出峰是什么原因,但是有很多杂峰?
DB21/T 2410-2015 养殖水体中氯霉素残留量的测定高效液相色谱串联质谱法
高效液相色谱串联质谱法分析烟草中15中农药残留烟草化学 2011年第7期
高效液相色谱-串联质谱法测定食品中20种氨基甲酸酯类农药残留
GB/T 30939-2014,化妆品中污染物双酚A的测定 高效液相色谱-串联质谱法,哪位手上有希望分享一下,照片,扫描件都可以,先谢谢了!

[align=left]超高效液相色谱–四极杆静电场轨道阱高分辨质谱仪(UPLC-Q/Orbitrap HRMS),在药物分析、代谢组学、脂质组学、安全风险物质筛查、环境分析等领域具有分辨率高、灵敏度高、准确性好的优势,在生物医药、食品安全、生命科学、环境健康、法医毒物、残留分析、产品检验等领域应用广泛。[/align][align=left][/align]
[align=center][font='宋体'][size=20px][color=#000000]超高效[url=https://insevent.instrument.com.cn/t/5p]液相色谱[/url]串联质谱测定花生及其制品中[/color][/size][/font][/align][align=center][font='宋体'][size=20px][color=#000000]16种真菌毒素[/color][/size][/font][font='宋体'][size=20px][color=#000000]实验过程出现的问题[/color][/size][/font][/align][font='宋体'][size=16px][color=#000000]运用超高效[url=https://insevent.instrument.com.cn/t/5p]液相色谱[/url]串联质谱测定花生及其制品中16种真菌毒素过程中[/color][/size][/font][font='宋体'][size=16px][color=#000000]色谱峰出现峰分叉和峰型不好现象。[/color][/size][/font][font='宋体'][size=16px][color=#000000]在实验中发现,离心参数的选择是油脂类制品前处理重要步骤,[/color][/size][/font][font='宋体'][size=16px][color=#000000]花生油脂的凝固点大致是0 ℃~3 ℃,[/color][/size][/font][font='宋体'][size=16px][color=#000000]经过反复实验,对比[/color][/size][/font][size=16px]离心[/size][size=16px]参数[/size][size=16px]得出[/size][size=16px]低温 2 ℃[/size][size=16px] [/size][size=16px]10 000 r/min离心10 min使脂类低温凝固,[/size][size=16px]可以[/size][size=16px]去除花生酱、花生油等样品中大量的脂类杂质,使提取液颜色变浅,澄清度更高,减少基质对测定的影响[/size][size=16px],[/size][size=16px]使峰[/size][font='宋体'][size=16px][color=#000000]型[/color][/size][/font][size=16px]得到很大改善。[/size][font='宋体'][size=16px][color=#000000]见[/color][/size][/font][font='宋体'][size=16px]图1[/size][/font][font='宋体'][size=16px][color=#000000]。[/color][/size][/font][align=left][img]https://ng1.17img.cn/bbsfiles/images/2023/10/202310241202333003_5694_4033901_3.jpeg[/img][/align][img]https://ng1.17img.cn/bbsfiles/images/2023/10/202310241202336208_9424_4033901_3.jpeg[/img][align=left][color=#333333]图[/color][color=#333333]1[/color][color=#333333] 16种真菌毒素混合标准溶液(MRM)色谱图[/color][/align][font='宋体'][size=16px][color=#000000] [/color][/size][/font]

超高效[url=https://insevent.instrument.com.cn/t/5p][color=#3333ff]液相色谱[/color][/url]-串联质谱法测定婴幼儿配方奶粉中叶酸的含量[align=center]户江涛[/align][align=center](黑龙江省农垦科学院测试化验中心,黑龙江 佳木斯 154007 )[/align]摘要:本实验建立了超高效[url=https://insevent.instrument.com.cn/t/5p][color=#3333ff]液相色谱[/color][/url]-串联质谱法建立了检测婴幼儿配方奶粉中叶酸含量的分析方法。试样经50℃热水溶解,超声提取,等电法去除蛋白后,在T[sub]3[/sub]色谱柱上以0.1%甲酸水和乙腈为流动相进行[url=https://insevent.instrument.com.cn/t/5p][color=#3333ff]液相色谱[/color][/url]分离,质谱检测采用电喷雾正离子化模式和多反应监测模式(MRM)。结果表明该方法在浓度范围内线性关系良好,相关系数(r)为0.9998,定量下限(LOQ)为10 [color=black]u[/color]g/100 g,加标回收率为83.7%~101%,相对标准偏差(RSD)为2.5%~5.1%(n=6)。本方法快速、灵敏、准确,特别适和大批量婴幼儿配方奶粉中叶酸含量检测。关键词:超高效[url=https://insevent.instrument.com.cn/t/5p][color=#3333ff]液相色谱[/color][/url]-串联质谱;婴幼儿配方奶粉;叶酸[align=center]Determination of Folic acid in infant formula by ultra performance liquid chromatography-tandem mass spectrometry[/align][align=center]HU Jiangtao[/align][align=center](Testing and Analysis Center of Heilongjiang Academy of Land Reclamation Sciences, Jiamusi 154007,China)[/align]Abstract:A method was developed for the determination of Folic acid in infant formula by ultra performance liquid chromatography-tandem mass spectrometry(UP[url=https://insevent.instrument.com.cn/t/Yp][color=#3333ff]LC-MS[/color][/url]/MS). The samples were disolved by 50℃ water,extracted by ultrasound,after protein was removed by Isoelectric method, Folic acid was separated on a Waters HSS T[sub]3[/sub] column with gradient elution with the mobile phase of 0.1% formic acid and acetonitrile, and finally detected by positive eletrospray ionization-mass spectrometry(ESI[sup]+[/sup]-MS/MS) in multiple reaction monitoring(MRM) mode. The results showed the linearity of Folic acid was good in the concentration range of 0.02~1 mg/L, and the correlation coefficient was 0.9998. The limit of quantification(LOQ) was 10 ug/100 g,the mean recovery was 83.7%~101%, and the relative standard deviation(RSD) was 2.5%~5.1%(n=6).This method is rapid, sensitive, accurate and suitable for simultaneous determination of Folic acid in infant formula.Key words: UP[url=https://insevent.instrument.com.cn/t/Yp][color=#3333ff]LC-MS[/color][/url]/MS infant formula Folic acid叶酸([font=helvetica][color=#333333]C[/color][/font][font=helvetica][sub][size=12px][color=#333333]19[/color][/size][/sub][/font][font=helvetica][color=#333333]H[/color][/font][font=helvetica][sub][size=12px][color=#333333]19[/color][/size][/sub][/font][font=helvetica][color=#333333]N[/color][/font][font=helvetica][sub][size=12px][color=#333333]7[/color][/size][/sub][/font][font=helvetica][color=#333333]O[/color][/font][font=helvetica][sub][size=12px][color=#333333]6 [/color][/size][/sub][/font]),[font=helvetica][color=#333333]又名维生素B[/color][/font][font=helvetica][sub][color=#333333]9[/color][/sub][/font][font=helvetica][color=#333333],[/color][/font]是一种水溶性维生素,在人类蛋白质合成、细胞分裂与生长过程中具有重要作用。叶酸缺乏可能会引起婴幼儿[font=helvetica][color=#333333]发生神经闭合不完全[/color][/font],从而导致神经性厌食症,叶酸虽然广泛存在于[font=helvetica][color=#333333]如菠菜等绿叶蔬菜及肝脏等动物源食品中,但在婴幼儿阶段(特别小于6个月婴儿)很长一段时间不能直接食用这些辅食,[/color][/font]这时婴幼儿配方奶粉便成为婴幼儿获取叶酸的重要途径,因此准确测定婴幼儿配方奶粉中叶酸含量有重要意义。目前食品中叶酸测定的国标方法《GB 5009.211-2022 食品安全国家标准 食品中叶酸的测定》为微生物法,该方法是将鼠李糖乳杆菌接种至含有叶酸的培养基中,培养一段时间后测定吸光度,利用在一定范围内叶酸含量与吸光度值符合某种规律而得到叶酸含量数值。该方法对菌种活力、培养条件、检验人员操作水平要求较高,检测周期较长,且容易受到基质干扰、检测结果重复性往往较差。因此,制定一种准确、高效、快速测定婴幼儿配方奶粉中叶酸的检测方法十分必要。[url=https://insevent.instrument.com.cn/t/5p][color=#3333ff]液相色谱[/color][/url]-串联质谱测定法具有前处理简单、分析速度快、抗基质干扰能力强等优点,能有效避免叶酸在长时间、繁琐的前处理及检测过程中损失,可以为奶粉中叶酸含量的测定提供一种有效的检测手段。本文建立的超高效[url=https://insevent.instrument.com.cn/t/5p][color=#3333ff]液相色谱[/color][/url]-串联质谱法测定奶粉中叶酸含量[color=black]的方法前处理过程简便、分析时间短、灵敏度高、抗干扰能力强,且用到的都是实验室常用试剂,实验成本较低,特别适用于大批量[/color]婴幼儿配方[color=black]奶粉中叶酸[/color]含量的检测。1 实验部分1.1 材料与试剂[color=black]叶酸(纯度[/color][font=宋体][color=black]≥[/color][/font][color=black]99%,Dr公司);乙腈、甲酸(色谱纯,Fisher公司);盐酸、氢氧化钠(优级纯,科密欧公司);0.2 um水系滤膜;实验用水为Millipore纯水仪制备。[/color]1.2 仪器与设备UPLC XEVO TQ-MS超高效[url=https://insevent.instrument.com.cn/t/5p][color=#3333ff]液相色谱[/color][/url]串联质谱仪(Waters公司);涡旋振荡器。1.3 [color=black]叶酸[/color]标准储备液的配置称取一定量叶酸[color=black]标准品[/color],用甲醇配置成质量浓度为100 ug/mL标准储备液,于-18℃冰箱保存,待用;临用前将溶液回温至室温,并吸取一定体积储备液用水逐级稀释成所需浓度的标准工作液。1.4 样品前处理准确称取2.00 g(精确到0.01 g) 婴幼儿配方奶粉试样于50 mL刻度离心管中,加入10 mL 50℃纯水使奶粉充分溶解,超声提取10 min,然后用2 mol/L盐酸调节试样pH值至1.7,静置5 min后用2 mol/L氢氧化钠调节试样溶液pH至4.5,用纯水定容至25 mL,在离心机中以10000 r/min离心10 min[color=black],取上清液[/color]过0.2 um水系微孔滤膜后供UP[url=https://insevent.instrument.com.cn/t/Yp][color=#3333ff]LC-MS[/color][/url]/MS分析测定。1.5 [url=https://insevent.instrument.com.cn/t/5p][color=#3333ff]液相色谱[/color][/url]及质谱条件[url=https://insevent.instrument.com.cn/t/5p][color=#3333ff]液相色谱[/color][/url]:色谱柱:Waters HSS [font=times new roman]T3(1.8 μm,50mm×2.1mm);柱温:35℃[/font];流速:[font=times new roman]0.3 [/font]mL/min;进样量:[font=times new roman]2[/font] [font=times new roman]μL;流动相A:乙腈;流动相B:0.1%的甲酸水溶液。梯度洗脱程序:0~0.5min,5% A;0.5~3. 0 min,5%~100% A;3. 0 ~4. 0 min,100%A,4 ~4.1min,100% A~5% A,4.1 ~5.0min 5% A。[/font]质谱:离子源:电喷雾离子源( ESI [sup]+[/sup] ) ;扫描方式:正离子扫描;检测方式:多反应监测( MRM);毛细管电压:3.0 kv;离子源温度:150℃;去溶剂气温度:500℃;去溶剂气流量:1000 L /h;定性、定量离子对及碰撞能量见表1。[align=center]表1叶酸的质谱参数[/align][table][tr][td][align=center]分析物[/align][/td][td][align=center]锥孔电压/V[/align][/td][td][align=center]母离子/(m/z)[/align][/td][td][align=center]子离子/(m/z) [/align][/td][td][align=center]碰撞能量/V[/align][/td][/tr][tr][td]叶酸[/td][td][align=center]18[/align][/td][td][align=center]442.3[/align][/td][td][align=center]295﹡[/align]176[/td][td][align=center]18[/align][align=center]30[/align][/td][/tr][/table]﹡为定量离子2 结果与讨论2.1 色谱质谱条件及前处理过程的优化流动相的选择:B族维生素在酸性条件下比较稳定,对比了酸性体系(0.1%甲酸水溶液)与甲醇、乙腈的流动相体系组合,发现叶酸在乙腈体系中响应值更高,故本研究采用0.1%甲酸水溶液+乙腈流动相体系。色谱柱的选择:比较了[font=宋体]Waters [/font]BEH C[sub]18[/sub](1.7 μm,50mm×2.1mm)和[font=宋体]Waters [/font]HSS T[sub]3[/sub](1.8 μm,50mm×2.1mm)两种不同填料的分析柱,实验时发现目标物在C[sub]18[/sub]上保留比T[sub]3[/sub]弱,考虑到若出峰太早可能造成奶粉中一些极性强的基质随目标物共流出,可能会干扰目标物测定,因此本方法采用了HSS T[sub]3[/sub]色谱柱。质谱参数优化:将1.0 mg/L 叶酸标准溶液直接注射到质谱中,在正离子模式下分别进行母离子和子离子全扫描,同时优化质谱条件,找到两对响应好高、干扰小的子离子对,最终确定的质谱条件见表1,相应的色谱质谱图见图1、图2。前处理过程优化:叶酸属于水溶性维生素,易于氧化,这个实验过程需在避光条件下进行。通过查阅相关文献的前处理方法,发现通过先酸化样品再用NaOH调节pH的等电法去除蛋白方式,可以得到很好的提取和净化效果,能大大减少奶粉中蛋白等大分子基质对目标物干扰;调节pH后的溶液产生了蛋白沉淀,需要用过滤或离心方式予以去除,通过实验比较了过滤和离心去除蛋白沉淀的效果,发现高速离心后得到的上清液明显更容易通过0.2 um水系滤膜,综合以上因素本实验最终采用了1.4的前处理方法。[align=center][img]https://ng1.17img.cn/bbsfiles/images/2023/10/202310041348335007_5097_1729077_3.png[/img][/align][align=center]图1 [color=black]叶酸[/color]标准溶液(1 ug/mL)MRM色谱图[/align][align=center][img]https://ng1.17img.cn/bbsfiles/images/2023/10/202310041348338570_9110_1729077_3.png[/img][/align][align=center]图2 奶粉样品中[color=black]叶酸[/color]MRM色谱图[/align][color=black]2.2 线性范围和定量限[/color][color=black]吸取不同体积的叶酸标准储备液(1.3),用[/color]纯水[color=black]分别配置不同浓度的[/color]上机标准溶液,以各自定量离子的峰面积为Y对应质量浓度X([color=black]m[/color]g/L)做标准曲线,得到的线性方程和相关系数见表2;以10倍信噪比(S/N)计算得到叶酸的定量下限,结果见表2。表2 叶酸标准溶液的线性方程、相关系数和定量下限(LOQ)[table][tr][td][align=center]分析物[/align][/td][td][align=center]线性范围/(mg/L)[/align][/td][td][align=center]线性方程[/align][/td][td][align=center]R[/align][/td][td][align=center]LOQ/(ug/100 g)[/align][/td][/tr][tr][td][align=center]叶酸[sub] [/sub][/align][/td][td][align=center]0.02~1[/align][/td][td][align=center]Y=12793X-56.759[/align][/td][td][align=center]0.9998[/align][/td][td][align=center]10[/align][/td][/tr][/table][color=black]2.3回收率和精密度[/color][color=black]选用[/color]不添加叶酸的奶粉样品作为基质进行加标。添加水平为:[color=black]20,100,200[/color][size=12px] ug/100g。[/size][color=black]每个[/color]水平重复6次,[color=black]同时做该奶粉的本底实验。[/color]按照1.4前处理方法处理后上机检测,回收率计算结果见表3。结果表明,该方法叶酸的平均回收率为83.7%~101%,相对标准偏差(RSD,n=6)为2.5%~5.1%,均满足实验要求。[align=center]表3 奶粉叶酸的加标回收率和相对标准偏差(n=6)[/align][table][tr][td][align=center]分析物[/align][/td][td][align=center]添加水平(ug/100g)[/align][/td][td][align=center]回收率/%[/align][/td][td][align=center]相对标准偏差/%[/align][/td][/tr][tr][td][align=center]叶酸[/align][sub] [/sub][/td][td][align=center]20[/align][align=center]100[/align][align=center]200[/align][/td][td][align=center]89.0±5.3[/align][align=center]95.3±2.7[/align][align=center]98.5±2.5[/align][/td][td][align=center]5.1[/align][align=center]3.2[/align][align=center]2.5[/align][/td][/tr][/table][color=black]2.4实际样品分析[/color][color=black]采用本方法随机抽取市售8批次婴幼儿配方奶粉,对其叶酸含量进行测定[/color],实测值、标示值及实测值与标示值比值见表4。结果表明,这些样品中叶酸实测含量均符合《GB 28050-2011 食品安全国家标准 预包装食品营养标签通则》中对维生素等营养成分的规定——婴幼儿配方奶粉中叶酸实际含量不应低于标示值的80%。[align=center]表4 实际样品中叶酸实测值与标示值比对结果[/align][table][tr][td][align=center]样品编号[/align][/td][td][align=center]实测值(ug/100g)[/align][/td][td][align=center]标示值(ug/100g)[/align][/td][td][align=center]比值[/align][/td][/tr][tr][td]1[sub] [/sub]2345678[/td][td][align=center]65[/align][align=center]156[/align][align=center]103.2[/align][align=center]57[/align][align=center]117[/align][align=center]86.5[/align][align=center]79[/align][align=center]101[/align][/td][td]81.3203230134106146198185[/td][td][align=center]1.25[/align]1.302.232.350.911.692.511.83 [/td][/tr][/table]3 结语本文建立了超高效[url=https://insevent.instrument.com.cn/t/5p][color=#3333ff]液相色谱[/color][/url]-串联质谱法(UP[url=https://insevent.instrument.com.cn/t/Yp][color=#3333ff]LC-MS[/color][/url]/MS)测定婴幼儿配方奶粉中[color=black]叶酸[/color]含量的分析方法。该方法具有较高的灵敏度、准确度和精密度,前处理快速、简单,特别适合大批量样品的检测。参考文献:[1] GB 5009.211-2016 食品安全国家标准 食品中叶酸的测定[s].[2] GB 28050-2011 食品安全国家标准 预包装食品营养标签通则[s].[3]张丽芳,张鑫,周鑫,等. 高效[url=https://insevent.instrument.com.cn/t/5p][color=#3333ff]液相色谱[/color][/url]法同时测定婴幼儿奶粉中7种水溶性维生素[J].[size=13px] [/size]食品工业,2022,43(01):[size=13px] [/size]277-280.[4]刘娜,陈大舟,汤桦,等. 婴幼儿奶粉中8种水溶性维生素的高效[url=https://insevent.instrument.com.cn/t/5p][color=#3333ff]液相色谱[/color][/url]同时测定[J].[size=13px] [/size]分析测试学报,2008,27(4):[size=13px] [/size]408-411.[5]严华,崔凤云,别玮,等. 超高效[url=https://insevent.instrument.com.cn/t/5p][color=#3333ff]液相色谱[/color][/url]-同位素稀释质谱法同时测定婴幼儿奶粉中10种水溶性维生素 [J].[size=13px] [/size]食品安全质量检测学报,2020,17(11):[size=13px] [/size]5871-5878.[6]郭建博,宋莉,牟霄,等.超高效[url=https://insevent.instrument.com.cn/t/5p][color=#3333ff]液相色谱[/color][/url]法快速测定复合维生素产品中的10种水溶性维生素 [J].[size=13px] [/size]食品安全质量检测学报,2017,8(5):[size=13px] [/size]1794-1799.[7]夏静,俞婧,孙磊,等.功能性饮料中9种水溶性维生素的HP[url=https://insevent.instrument.com.cn/t/Yp][color=#3333ff]LC-MS[/color][/url]-MS同步检测技术 [J]. 食品科学,2014,35(12): 196-199.[/s][/s]
摘要:本文建立了蔬菜和水果中赤霉素残留的高效液相色谱-串联四极杆质谱联用测定方法。该方法经50%乙腈水溶液(pH值2.5)提取样品,以ZORBAX SB-Aq柱(2.1mm×150.0mm,3.5μm)分离,流动相为0.1%甲酸水溶液和甲醇(体积比为60∶40),电喷雾正离子MRM模式检测。该方法的检出限2.0μg/L,方法定量下限10μg/kg,线性范围2.0~10.0μg/L,加标回收率90.1%~102.3%,相对标准偏差为4.07%。
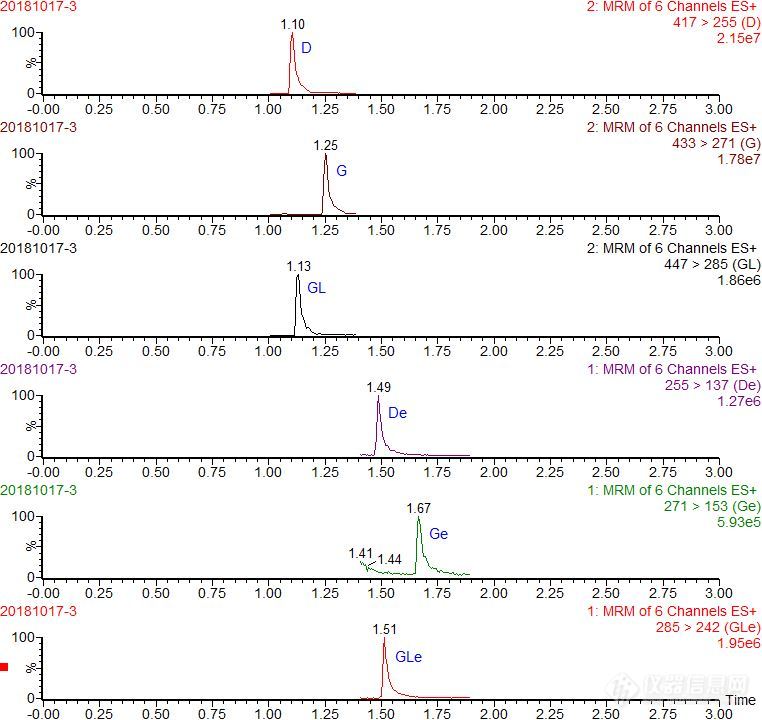
[align=center][b]超高效液相色谱-串联质谱法测定大豆中大豆异黄酮的含量[/b][/align][align=center]户江涛[/align][align=center](农业农村部豆类产品质量安全风险评估实验室(佳木斯),黑龙江省农垦科学院测试化验中心,黑龙江佳木斯 154007 )[/align]摘要:采用超高效液相色谱-串联质谱法建立了检测大豆中大豆异黄酮含量的分析方法。试样经90%甲醇水提取后,6种大豆异黄酮在C[sub]18[/sub]色谱柱上以0.1%甲酸水溶液和乙腈为流动相,进行液相色谱分离;质谱检测采用电喷雾正离子化模式和多反应监测模式(MRM)。结果表明,6种大豆异黄酮分别在0.01~0.5 mg/L(D、GL、G)和0.002~0.1 mg/L(De、GLe、Ge)范围内线性关系良好,相关系数(R)为0.9993~0.9998,定量限(LOQ)为0.0001 g/kg。在大豆空白样品添加浓度分别为0.01、0.05、0.2 g/kg(De、GLe、Ge)和0.2、1、2 g/kg(D、GL、G),6种大豆异黄酮的平均回收率为86.6%~96.2%,相对标准偏差(RSD)为1.07%~5.93%(n=6)。本方法简便、灵敏、抗干扰,适用于大豆中大豆异黄酮含量检测。关键词:超高效液相色谱-串联质谱;大豆;大豆异黄酮[align=center]Determination of soybeanisoflavone in soybean by ultra performance liquid chromatography-tandem massspectrometry[/align][align=center]HU Jiangtao[/align][align=center]([i]Laboratory of Qualityand Safety Risk Assessment for Soybean products, Ministry of Agriculture andRural Affairs, Testing and Analysis Center of Heilongjiang Academy of LandReclamation Sciences, Jiamusi 154007,China[/i])[/align][b]Abstract:[/b]A methodwasdeveloped for the determination of soybeanisoflavone in soybean by ultra performance liquid chromatography-tandem massspectrometry(UP[url=https://insevent.instrument.com.cn/t/Yp][color=#3333ff]LC-MS[/color][/url]/MS). The samples were extracted by 90% methanol-water, then 6 soybean isoflavones were separated on aWaters BEH C[sub]18[/sub] column with gradient elution with the mobile phase of0.1% formic acid and acetonitrile, and finally detected by positive eletrosprayionization-mass spectrometry(ESI[sup]+[/sup]-MS/MS) in multiple reactionmonitoring(MRM) mode. The results showed the linearities of 6 soybean isoflavones were good in the concentrationrange of 0.01~0.5 mg/L(D、GL、G)and 0.002~0.1 mg/L(De、GLe、Ge), the correlation coefficients were 0.9993~0.9998. The limitof quantification(LOQ) of soybean isoflavone was 0.0001 g/kg. At the spiked levels of 0.01、0.05、0.2 g/kg(De、GLe、Ge)and 0.2、1、2 g/kg(D、GL、G) in the blank soybean samples, the mean recovery of soybeanisoflavone was 86.6%~96.2%, andthe relative standard deviation(RSD) was 1.07%~5.93%(n=6).This method is simple,sensitive, anti-jamming and suitable for simultaneous determination of soybean isoflavone in soybean.[b]Key words: [/b]ultra performance liquid chromatography-tandem massspectrometry (UPLC-MS/MS) soybean soybean isoflavone大豆异黄酮(soybean isoflavone)是一族化合物的统称,是大豆植物体内的一种次生代谢产物,是大豆主要活性成分之一,其母核为3-苯并吡喃酮,主要包括大豆苷、大豆黄苷、染料木苷及其相应苷元[sup][/sup]。研究表明,大豆异黄酮除具有天然抗氧化作用外[sup][/sup],还具有降低胆固醇含量、预防多种癌症及改善妇女更年期综合征等多方面生物功效[sup][/sup]。大豆异黄酮主要存在于大豆籽实中,其总含量约为0.4~5 g/kg,其中大豆苷、大豆黄苷和染料木苷这三种含量约占总量的97%~98%,而其对应的苷元含量仅占2%~3%左右[sup][/sup]。目前,大豆异黄酮的检测方法主要有高效液相色谱法(HPLC)[sup][/sup]、[url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]法[sup][/sup]、紫外分光光度法[sup] [/sup]、质谱法(HP[url=https://insevent.instrument.com.cn/t/Yp][color=#3333ff]LC-MS[/color][/url]/MS[sup][/sup])等。紫外分光光度法[sup] [/sup]只能测定大豆异黄酮总量,且灵敏度不高;[url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]法[sup][/sup]需要对异黄酮进行衍生,前处理复杂;目前,大豆异黄酮检测现有的国家标准GB/T 26625-2011[sup] [/sup]采用的是高效液相色谱法(HPLC),在实际检测过程中发现,由于紫外检测器灵敏度不高,存在个别样品中异黄酮相应苷元检测不到的情况;同时大豆提取液中含有蛋白、脂肪等杂质影响色谱柱柱效,以至于不能满足分离度要求,严重干扰低含量组分峰面积积分定量。而[url=https://insevent.instrument.com.cn/t/Yp][color=#3333ff]LC-MS[/color][/url]/MS法质谱检测器灵敏度高,通过选定大豆异黄酮的特征离子,能有效去除上述杂质干扰,定量更加准确可靠。目前,国内外采用[url=https://insevent.instrument.com.cn/t/Yp][color=#3333ff]LC-MS[/color][/url]/MS法检测大豆中大豆异黄酮含量的文献很少[sup][/sup]。本文对大豆中大豆异黄酮检测的前处理方法借鉴GB/T 26625-2011[sup][/sup],提取液改用UP[url=https://insevent.instrument.com.cn/t/Yp][color=#3333ff]LC-MS[/color][/url]/MS测定。该方法前处理过程简便、灵敏度高、分析时间短、抗干扰能力强,适用于大批量大豆样品中大豆异黄酮含量的检测。[b]1 实验部分[/b]1.1 材料与试剂大豆苷(daidzin,记为D,以下同)、大豆黄苷(glycitin,GL)、染料木苷(genistin, G)、大豆素(daidzein,De)、大豆黄素(glycitein, GLe)、染料木素(genistein,Ge)(纯度≥99%,Dr.Ehrenstorfer公司);甲醇、乙腈、甲酸(色谱纯,Fisher公司);实验用水为Millipore纯水仪制备。1.2 仪器与设备Acquity UPLC型超高效液相色谱仪(Waters公司);XEVO TQ-S三重四级杆质谱仪(Waters公司);KQ-500DE型超声波仪(昆山市超声仪器有限公司);涡旋混合器(IKA公司);CR21GⅢ型高速离心机(HITACHI公司)。1.3 大豆异黄酮标准储备液的配置分别称取适量的D、GL、G、De、GLe、Ge标准品,用甲醇配置成质量浓度为1mg/mL标准储备液,于-18℃冰箱保存(有效期6个月),待用;使用时用10%甲醇水逐级稀释成所需浓度的混合标准工作液,现用现配。1.4 样品前处理提取:称取粉碎均与后的试样1.0g(精确到0.01g)于50mL聚乙烯离心管中,加入10.0 mL90%甲醇水,涡旋混合30 s后置于60℃超声波清洗器中提取30 min,在离心机中以15000 r/min离心5 min,将上清液转移至100 mL容量瓶中,残渣再加入10.0 mL90%甲醇水溶液按上述步骤提取后,合并两次上清液于100 mL容量瓶中,用10%甲醇水溶液定容至刻度,摇匀。a)De、GLe、Ge的测定:取1 mL过0.22um有机系微孔滤膜,供UPLC/MS/MS分析测定;b)D、GL、G的测定:由于D、GL、G含量较高,需要将a)中过完滤膜的待测液用10%甲醇水稀释50倍后,供UPLC/MS/MS分析测定。1.5 液相色谱及质谱条件液相色谱:色谱柱:Waters BEH C[sub]18[/sub](1.7 μm,50mm×2.1mm);柱温:30℃;流速:0.5 mL/min;进样量:1μL;流动相A:乙腈;流动相B:0.1%的甲酸水溶液。梯度洗脱程序:0~0.5min,10% A;0.5~3. 0 min,10%~100% A;3. 0 ~4. 0 min,100%A,4 ~4.1.1min,100% A~10% A,4.1 ~5.0min 10% A。质谱:离子源:电喷雾离子源( ESI [sup]+[/sup] ) ;扫描方式:正离子扫描;检测方式:多反应监测( MRM);毛细管电压:3.2 kv;离子源温度:150℃;去溶剂气温度:500℃;去溶剂气流量:1000 L /h;定性、定量离子对及碰撞能量见表1。[align=center]表1大豆异黄酮的质谱参数[/align][align=center]Table 1 MRM parameters of soybean isoflavone[/align] [table][tr][td] [align=center]Analyte[/align] [/td][td] [align=center]Cone/V[/align] [/td][td] [align=center]Parent ion/(m/z)[/align] [/td][td] [align=center]Daughter ion/(m/z)[/align] [/td][td] [align=center]Collision energy/V[/align] [/td][/tr][tr][td] [align=center]D[/align] [align=center] [/align] [/td][td] [align=center]30[/align] [align=center] [/align] [/td][td] [align=center]417[/align] [align=center][sup] [/sup][/align] [/td][td] [align=center]255﹡[/align] 137[/td][td] [align=center]27[/align] [align=center]18[/align] [/td][/tr][tr][td] [align=center]G[/align] [align=center] [/align] [/td][td] [align=center]30[/align] [align=center] [/align] [/td][td] [align=center]433[/align] [align=center][sup] [/sup][/align] [/td][td] [align=center]271﹡[/align] 153[/td][td] [align=center]21[/align] [align=center]50[/align] [/td][/tr][tr][td] [align=center]GL[/align] [align=center] [/align] [/td][td] [align=center]30[/align] [align=center] [/align] [/td][td] [align=center]447[/align] [align=center][sup] [/sup][/align] [/td][td] [align=center]285﹡[/align] 270[/td][td] [align=center]25[/align] [align=center]46[/align] [/td][/tr][tr][td] [align=center]De[/align] [align=center] [/align] [/td][td] [align=center]25[/align] [align=center] [/align] [/td][td] [align=center]255[/align] [align=center][sup] [/sup][/align] [/td][td] [align=center]137﹡[/align] 181[/td][td] [align=center]30[/align] [align=center]26[/align] [/td][/tr][tr][td] [align=center]Ge[/align] [align=center] [/align] [/td][td] [align=center]25[/align] [align=center] [/align] [/td][td] [align=center]271[/align] [align=center][sup] [/sup][/align] [/td][td] [align=center]153﹡[/align] 215[/td][td] [align=center]30[/align] [align=center]25[/align] [/td][/tr][tr][td] [align=center]GLe[/align] [align=center] [/align] [/td][td] [align=center]25[/align] [align=center] [/align] [/td][td] [align=center]285[/align] [align=center][sup] [/sup][/align] [/td][td] [align=center]242﹡[/align] 168[/td][td] [align=center]27[/align] [align=center]35[/align] [/td][/tr][/table]﹡quantitativeion[b]2 结果与讨论[/b]2.1 色谱及质谱条件的优化流动相的选择:对比了酸性体系(0.1%甲酸水溶液)与非酸性体系(纯水、乙酸铵溶液)分别与甲醇、乙腈的流动相体系组合,结果发现目标物在酸性体系中比非酸性体系响应更高、峰形更好;同时大豆提取液中含有蛋白、脂肪等杂质可能会残留在色谱柱上,影响色谱柱的使用寿命,而乙腈比甲醇体系洗脱能力更强,可以有效去这些杂质。综合考虑目标物信号强度、色谱分离效果以及除杂等因素,本研究采用0.1%甲酸水溶液+乙腈流动相体系。质谱的选择:根据6种大豆异黄酮的分子量,用10%甲醇水配置1.0 mg/L 大豆异黄酮标准溶液直接注射到质谱中,在正离子模式下分别对各种组分进行母离子及对应子离子全扫描,最终确定的质谱条件见表1。2.2 质谱法(UP[url=https://insevent.instrument.com.cn/t/Yp][color=#3333ff]LC-MS[/color][/url]/MS)与色谱法(HPLC)的比较国家标准《GB/T 26625-2011粮油检验大豆异黄酮含量测定高效液相色谱法》[sup][/sup]中规定的大豆异黄酮检测方法为HPLC法。对同一大豆样品分别采用本文UP[url=https://insevent.instrument.com.cn/t/Yp][color=#3333ff]LC-MS[/color][/url]/MS法(MRM色谱图见图1、2)和GB/T 26625 HPLC法检测,结果表明这两种方法测定的大豆异黄酮总含量值基本一致。由于De、GLe、Ge这三种苷元在大豆中含量很低,用HPLC法检测时,紫外检测器灵敏度不高,存在个别样品中上述三种组分检测缺失的情况;同时在实际大批量样品检测中发现,随着进样次数的增加,色谱柱柱效下降,大豆提取液中存在的蛋白、脂肪等杂质对含量低的目标物峰干扰越来越大,定量困难。研究发现,同浓度的大豆异黄酮在质谱检测器上的响应值要远远超过紫外检测器,同时质谱法可以通过选定大豆异黄酮的特征离子,有效地去除杂质的干扰,其目标物分离度不受色谱柱进样次数增加的影响,定量更加准确可靠。[align=center][img=,690,651]https://ng1.17img.cn/bbsfiles/images/2019/08/201908050912587968_4111_3299836_3.jpg!w690x651.jpg[/img][/align][align=center]图1 大豆异黄酮标准溶液(0.01mg/L)MRM色谱图[/align][align=center]Fig.1 MRM chromatograms of soybean isoflavone standard solution at 0.01 mg/L[/align][align=center][/align][align=center][img=,690,653]https://ng1.17img.cn/bbsfiles/images/2019/08/201908050913342201_5843_3299836_3.jpg!w690x653.jpg[/img][/align][align=center]图2 大豆样品中大豆异黄酮MRM色谱图[/align][align=center]Fig.2 MRM chromatograms of soybean isoflavone in soybean[/align]2.3线性范围和定量限吸取不同体积的大豆异黄酮标准储备液(1.3),用10%甲醇水分别配置0.002、0.005、0.01、0.05、0.1(De、GLe、Ge)和0.01、0.05、0.1、0.2、0.5(D、GL、G)的大豆异黄酮上机混合标准溶液,以各自定量离子的峰面积为Y对应质量浓度X(mg/L)做标准曲线,得到的线性方程和相关系数见表2。结果表明,大豆异黄酮标准溶液在各自浓度范围内线性良好,相关系数R为0.9993~0.9999。以10倍信噪比(S/N)计算,大豆异黄酮上机液最低定量浓度为0.001 mg/L,通过公式(1)计算得到大豆中大豆异黄酮含量,最终确定本方法大豆异黄酮的定量限(LOQ)为0.0001 g/kg。糠氨酸质量分数计算公式:[align=center][img=,207,87]https://ng1.17img.cn/bbsfiles/images/2019/08/201908050915166414_5621_3299836_3.jpg!w207x87.jpg[/img] ………………(1)[/align] 式中:X为试样中大豆异黄酮含量,以g/kg计;C为大豆异黄酮上机浓度(mg/L);V为定容体积(V=100)。表2 大豆异黄酮标准溶液的线性方程和相关系数[align=center]Table 2 Linear equation and correlation of soybean isoflavone in 10% methanol-water standard solutions[/align] [table][tr][td] [align=center]Analyte[/align] [/td][td] [align=center]Linear range/(mg/L)[/align] [/td][td] [align=center]Linear equation[/align] [/td][td] [align=center]R[/align] [/td][td] [align=center] [/align] [/td][/tr][tr][td] [align=center]D[/align] [align=center]GL[/align] [align=center]G[/align] [align=center]De[/align] [align=center]GLe[/align] [align=center]Ge[/align] [/td][td] [align=center][sup]0.01~0.5[/sup][/align] [align=center][sup]0.01~0.5[/sup][/align] [align=center][sup]0.01~0.5[/sup][/align] [align=center][sup]0.002~0.1[/sup][/align] [align=center][sup]0.002~0.1[/sup][/align] [align=center][sup]0.002~0.1[/sup][/align] [/td][td] [align=center]Y=2393.6x+479.38[/align] Y=1885x+139.66 [align=center]Y=1470.9x+187.97[/align] [align=center]Y=4287.9x+442.79[/align] [align=center]Y=3521.7x-103.62[/align] [align=center]Y=1993x+122.79[/align] [/td][td] [align=center]0.9995[/align] [align=center]0.9999[/align] [align=center]0.9993[/align] [align=center]0.9998[/align] [align=center]0.9997[/align] [align=center]0.9998[/align] [/td][td] [/td][/tr][/table]2.4回收率和精密度大豆中De、GLe、Ge含量较低,而D、GL、G含量较高,故本方法准确度实验分为高低浓度梯度组进行加标。称取大豆试样1.00 g,分别添加0.01、0.05、0.2 g/kg(De、GLe、Ge)和0.2、1、2 g/kg(D、GL、G),每个水平重复6次,同时做该大豆的空白本底实验。按照1.4前处理方法处理后上机检测,计算回收率(扣除空白),结果表明:不同添加浓度下,De、GLe、Ge的平均回收率为91.7%~96.2%,相对标准偏差(RSD,n=6)为2.78%~5.93%;D、GL、G的平均回收率为86.6%~93.8%,相对标准偏差(RSD,n=6)为1.07%~3.77%。[b]3 结语[/b]本文建立了超高效液相色谱-串联质谱法(UP[url=https://insevent.instrument.com.cn/t/Yp][color=#3333ff]LC-MS[/color][/url]/MS)测定大豆中大豆异黄酮含量的分析方法。该方法灵敏度高,线性范围宽,能同时覆盖大豆中多梯度浓度大豆异黄酮组分含量的测定。同时该方法具有较高的准确度和精密度,前处理步骤简单,分析速度快,可有效避免由于色谱柱柱效下降对最终检测结果的影响,特别适合大批量样品的检测。田娟娟, 宋宏哲, 张飞, 等. 水剂法纯化大豆异黄酮的研究. 大豆通报, 2005, 6:19-22. Hagen M K, Ludke A, Araujo A S, et al.Antioxidant characterization of soy derived products in vitro and the effect ofa soy diet on peripheral markers of oxidative stress in a heart disease model .Canadian Journal of Physiology and Pharmacology, 2012,90(8):1095-1103. 徐春华, 张治广, 谢明杰, 等. 大豆异黄酮的抗氧化和抗肿瘤活性研究研究 . 大豆科学, 2010, 29(5): 870-873. 李俏俏, 王清路, 薛金艳, 等. 大豆异黄酮对绝经女性血清中脂类物质的影响的研究 . 大豆科学, 2009, 28(1):172-174. 胡润芳, 张玉梅, 陈宇华, 等. 大豆异黄酮含量的初步研究. 东南园艺, 2017, 6:9-11. 刘琴, 朱媛媛, 白兴梁. 不同种类大豆中大豆异黄酮含量及抗氧化性比较. 北京工商大学学报(自然科学版), 2012, 30(6): 45-51. 袁凤杰, 姜莹, 董德坤, 等. 中国大豆核心种质异黄酮含量分析.中国粮油学报, 2011, 26(2):5-8. Tepavcevic V, Atanackovic M,Miladinovic J,et al. Isoflavone composition,total polyphenolic content,and antioxidant activity in soybeans of different origin. MedFood,2010,13(3):657-664 GB/T 26625-2011《粮油检验大豆异黄酮含量测定高效液相色谱法》. Liggins J,Bluck J C. Deidzein and genistein content of fruits and nuts. Journal ofNutritional Biochemistry,2000,11(6):326-331. 鞠兴荣, 袁建, 汪海峰. 三波长紫外分光光度法测定大豆异黄酮含量的研究. 食品科学, 2001, 22(5):46-48.







 我要推广仪器
我要推广仪器
 下载APP
下载APP