根据GB17592-2011,纺织品偶氮检测中的定量过程中,请问大家采用的是“混标液+多种内标液”,还是“单标+一种内标液”,若是前者,具体是按照什么比例配制?如果是后者,具体是按照什么比例加配制?
内标液浓度是4.07,但是已经用完了,现在从新配置的内标液浓度是4.1 ,想问一下,校正因子是否需要重新计算,为什么?还是直接把以前的4.07 改为4.1就可以了?
http://support.lsca-china.com.cn/demo/agilentbbs/Skins/Default/emot/em01.gif您好。单位新安装了ICP-MS,是7700.我看了几遍工程师给的现场教材,有几个问题我还是没弄明白,所以特意在这里向各位大侠请教一下:1.我看到操作说明书上测样品时,既需要配标准曲线,也需要添加内标。我在这里就比较疑惑了:因为我之前是做色谱的,标准曲线=外标法,加内标=内标法,通常我们在色谱里用外标法定量就不需要添加内标的,用内标法定量就不需要配制工作曲线的。。。。。。但是在ICP-MS里面既用外标法,也添加内标,我就搞糊涂了,想不明白。。。。。还请各位大侠指导一下,为什么要这样做,这里面添加的内标起到什么作用,它参加最后的定量计算吗??最后的定量是通过标准曲线,还是内标定量??关于内标,我还有一个疑惑:教材上说内标溶液中包含6LI,Sc,Ge,Rh,In,Tb,Lu,Bi这8种元素。请问一下,如果我想校正某一个待测元素,比如As,这8个内标元素都参与到对As的校正过程里面去吗??如果是,请问一下各个元素在校正过程中的相应权重是多少??此外,是否所有待测元素的校正用这8种元素就足够了??还有关于虚拟内标的问题,请问它的原理是什么??还有它的准确性怎么样??2.关于标液,教材中是这样来配制标准溶液的。• 用 1%HNO3 稀释 Calibration Verification Standard ( Part # 5183-4682) 或 Environmental Calibration Standard (Part # 5183-4688, 1000ppm Fe, K, Ca, Na, Mg 和 10ppm Ag, As, Se, Cd, Pb, Ni, Cu, Zn 等) 到 1000 和 500 倍,得到 STD1 和 STD2。• 准备 1ug/L 和 2 ug/L Hg在 1% HNO3中,作为 STD3 和 STD4。• 空白 1% HNO3 为 STD0。Question:空白溶液我可以理解,但是“STD1、STD2”和“STD3、STD4”中所含有的元素是不一样的。。。。。。这样配制的标准曲线。我百思不得其解。。。。。。。。标准曲线各个点里面含有的元素种类不是应该都一样的吗??问题比较多,还请各位不吝赐教~~。O(∩_∩)O谢谢http://support.lsca-china.com.cn/demo/agilentbbs/Skins/Default/emot/em01.gifhttp://support.lsca-china.com.cn/demo/agilentbbs/Skins/Default/emot/em01.gif
有哪位大侠能告之俺白酒色谱分析中两内标的详细配制方法.谢谢!!
大家用内标物是买纯品自己配制还是买标液?如果自己配制是称量纯品重量还是吸取体积?
液相色谱测定苯甲酸时,配制苯甲酸标液,标准品是苯甲酸固体,可是苯甲酸在水中几乎不溶解,要怎么配制苯甲酸标液,又能不产生误差?求解决。
本菜鸟最近在用GC-MS测定烟包中18种光引发剂 按照标准使用内标法测定 标准中系列标准工作溶液配制如下:分别从母液(100ug/mL)中准确移取0.2mL、0.5mL、1.0mL、 2.0mL、 5.0mL于5个50mL容量瓶中,加入200uL内标溶液(内标浓度1mg/mL),用溶剂定容待用。 则配制的系列标准工作溶液中光引发剂含量为20ug、50ug、100ug、200ug、500ug;内标含量为200ug。绘制标准工作曲线是以光引发剂与内标峰面积比为纵坐标,以各标准工作溶液中光引发剂与内标的质量比为横坐标(最终结果要求是质量单位)。我困惑的地方在于 配制系列标准液时候, 如果以质量比做横坐标的话,那是不是只要保证系列标准工作溶液中目标物的质量不变,定容到任何体积都可以,不一定非得定容到50mL?(也就是保证质量一致但不保证浓度一致)可是如果体积不一样的话进入检测器的目标物的量肯定是不一样的啊,所以是不是这里的的质量比只是因为结果要求为质量,所以才用质量比做横坐标的,实际上内标法要求的还是浓度比?该方法样品前处理时是把样品放在40mL的溶剂中,加入200ug内标,然后进行后边的处理。我猜想系列工作溶液定容到50mL是为了标液环境尽量贴近样品处理时的环境?不知自己的考虑方向对不对从另外一个角度考虑,如果保证浓度不变的话,在建标曲输入横坐标的量时候,岂不是在保证目标物和内标比例不变的情况下,可以随便写目标物的质量范围?例如按照原标准方法中的系列标液,我可以建立第一个图的标准曲线;从这一系列标液中分别吸取5mL再作为标液,则可以建立第二个图的标准曲线。区别就是加的内标的量不一样了,我只要在样品处理时,根据曲线的不同,加入相应的内标量就可以。 顺着这个思路,就可以在只配一次系列标准液的情况下,建立任意质量范围的标准曲线了。因为从烟草局买回来的标液就是浓度跟方法里的标液浓度一样,但每一级只给了1mL,所以由此引发了上述思路,不知道该怎么做标曲了。。。感觉自己陷入了一个逻辑怪圈,绕不出来,请有这方面经验的大神帮忙指导一下,小女子在此先谢过啦![img]http://ng1.17img.cn/bbsfiles/images/2018/03/201803121019315704_6401_3062055_3.jpeg[/img]
由于我刚刚接触这方面的工作,好多都不是很了解,我们这配IC标液时用到的试剂有碳酸钠和碳酸氢钠,向各位请教一下IC标液的配制方法,谢谢
[color=#454545]液相色谱测定苯甲酸时,配制苯甲酸标液,标准品是苯甲酸固体,可是苯甲酸在水中几乎不溶解,要怎么配制苯甲酸标液,又能不产生误差?[/color]
电化学打标液成分与配制方法,,通过电流后,能在不锈表面形成文字或图案的“电蚀液”,求教这种液体的配方。
各位大侠, 本人现在要配含有一定浓度内标的标准溶液,有些搞不太清楚的问题?情况是这样的: 现有内标A ,标准物B、C。我先称一定量的内标A,然后用溶剂稀释,配成一定浓度的内标溶液D , 接着我称一定量的B、C标准物,稀释用的溶剂为刚配好的含有内标的溶液(也就是上面的D溶液),然后逐级稀释溶液,稀释过程中用的稀释剂都为D溶液,请问我最后配出来的各级标液里的内标浓度都相同吗?若是错了,请问该如何配才能使各级标液里的内标浓度都相同?
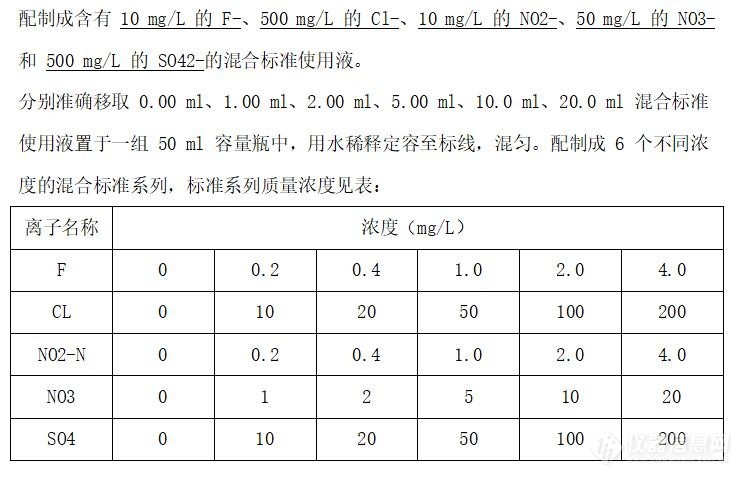
[img=,690,443]https://ng1.17img.cn/bbsfiles/images/2022/06/202206281712462900_7595_5273478_3.jpg!w690x443.jpg[/img][size=18px]如图,CL和SO4配置的浓度比标准上的高了5倍,结果做出来的曲线相关系数只有1个9。F和NO3比标准上的高了2倍,但相关系数结果都是>0.995的。是否色谱柱不适用浓度太高的标液?[/size]
标准里提到的配制不同浓度标准物质系列,然后同体积进样的方法。如果把这种方法的思维反过来,做法也同样也反过来,即标准物质的浓度是相同的,量取的标准物质的体积是不同的。比如说,用同一种标液分别取2μL、1.6μL、1.2μL、0.8μL、0.4μL等5种不同的体积的同一浓度的标准物质,这个浓度一般可以使用标准物质的原始浓度,它们所含的标准物质的质量就不同,可以根据标物的浓度计算各个浓度点所含标物的质量,将五个体积的标物打进色谱仪就可以得到相应的五个峰面积。根据标物的质量和峰面积就可以做出标准曲线。
请问离子色谱的标准溶液怎么配制?
由于急需砷标液,三氧化二砷太贵,现成的砷标液不好买!只有纯的砷粉,不知道有没有在普通实验室可以用砷粉配制砷标液的方法!谢谢!!!
刚接触饲料中氨基酸的检测,打算用OPA柱前衍生—液相色谱法来做。购买了氨基酸的固体粉末,每一种氨基酸约0.1g。用0.1%的盐酸溶液来配制?但是不知道是稀释到什么程度,看一些资料的氨基酸结果浓度的表示方式都是** %,标准溶液是** pmol/L。因为之前一直都是做食品的,用的单位都是mg/kg或者mg/L。想不明白应该如何配制配液。氨基酸检测一般是用单标定量,还是曲线定量?外标?内标?谢谢!
为什么相同的含量的标液,不同的标准采用不同的配制方法?
ICP测试标液的配制?谁有方法啊,能不能共享一下??
配制农药标液时,用丙酮定容,用10mL容量瓶配制与用25mL容量瓶配制,哪个准确度更高些,从理论上来讲,浓度不一样峰面积应该不一样,但最近我用这样的方法配制标液,峰面积却一样。1、吸400uL(浓度为10ug/mL)毒死蜱到25mL容量瓶中,定容至刻度,浓度为0.16ug/mL,峰面积为5900。2、吸200uL(浓度为10ug/mL)毒死蜱到10mL容量瓶中,定容至刻度,浓度为0.20ug/mL,峰面积为5900。移液用同一支移液枪(100-1000uL),在天平上校验过200uL,还是准确的。这是为什么?
如题,请问标液该如何配制,二甲醚易挥发,浓度不稳定。我们样品中甲醇约60%,二甲醚含量在0.6%作用,试问二者浓度差别这么大,用多大的分流比合适,能在同一台色谱仪分析吗?
公司刚买了PE 的全套ATD-GC-MS,要做室内空气的苯系物的测定,没有买苯系物的混合标液,只是买了苯,甲苯,二甲苯等的色谱纯试剂,请问如何将这些配制成混合标液呢?我看了色谱纯的甲苯的参数,纯度=99.8%,我用GC-MS做了一下,纯度不错,没发现其他苯系物的杂峰.请问是不是就把这个试剂看成是纯的甲苯,用他来直接配制混标?如果是的话,是不是用微量取样器量取一定体积,然后乘以甲苯密度,得到甲苯的质量,再除以体积得到浓度(xx ug/mL)呢?另外大家用什么来稀释苯系物呢?是用二硫化碳吗?配的混合标液中各组分的浓度是多大?最后做标准曲线的时候,是不是直接用微量进样器量取一定体积后直接注入采样管呢(sample tube)?然后再用氮气吹一会采样管?我的是ATD进样(冷阱捕集后再二级脱附).老板催的紧,下了死命令,要下周一前出报告,不然可能就要被炒了,好担心啊,请大家帮帮忙吧`~~~~谢谢谢谢~~~~~~~~[em63] [em49]
有谁知道液相测试硝基芳烃和硝基胺,做曲线时候标液配制的梯度是那几个浓度?给介绍一下经验
为啥同位素稀释质谱法测定时同等浓度标液和内标峰面积差了近一倍?配制过程没有好大问题,这是什么情况,这种情况正常么??
请教各位老师,一共有9组不同成分的铝钛硼合金待测,称样量0.1000-0.1300g之间,查阅文献前处理方法有的是稀盐酸反应,再加硝酸;有的是加入氢氟酸(含钛的原因),不知道哪种方法准确,具体加入比例有一般经验吗? 关于标液的配置如何配置比较好,没有固体标样,只有Si和B的1000ppm的单标液和含有Ti Zn Fe Cu Mn Mg等近20种元素的混标液,为了减轻基体效应,配了酸溶99.9995%Al基体溶液10000ppm,与单标和混标液混合制作工作曲线,根据上述称样量和以下几个主要待测元素的含量,稀释到多少毫升较好配合所配标液线性范围? 测试合金中的6-7个元素含量,主要待测元素含量如下:Si元素含量0.04%,Zn元素含量0.03%,Ti 3-5%左右,B 1%左右。
有内标法具体操作方法吗?如何配制系列标准液?是将相同量的内标物加到不同浓度的样品中吗?
想请教一下各位大神们 偶氮我们买的是1ml的混标浓度1000ug/ml 三种内标买的也是1ml 1000ug/ml浓度的标物我现在的内标法做法是先把5个梯度的标准溶液配制好定容到10ml容量瓶后 再把混合内标配制好浓度为50ug/ml 然后吸1ml标液加20ul标液到[url=https://insevent.instrument.com.cn/t/bp][color=#3333ff]GCMS[/color][/url]进样品里 上机测试做样品也是每个样品吸1ml加20ul标液后上机。总感觉这样子做有点不对各位老师们你们是怎么做的偶氮曲线和样品 主要是内标液的配制和加法要怎么做 浓度低也不好配 内标法没搞明白 请指导小弟一下 先在这谢谢了
请问各位,温度对配制标液影响大吗?因而建立的方法有影响吗?
如题:为啥同位素稀释质谱法测定时同等浓度标液和内标峰面积差了近一倍?配制过程没有好大问题,这是什么情况,这种情况正常么??
请教各位大神! 我们在进行[url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]方法验证时,遇到问题:1.检测限和定量限有没有必要做? 2.线性如何做,有必要从定量限开始做吗? 2个问题参考以下方法 具体方法介绍如下:内标法 待测物质是A物质 内标物是B物质 产品A物质的规格限为3.5~5.4(备注:检测结果低于3.5为不合格,高于5.4也不合格) 线性我们做的是规格限的60%~150%(做线性时(只用A物质标液,没有用到内标物,单独考察A物质峰面积和浓度关系),是否合理。

俺从事色谱分析和管理工作十余年拉,看见好多板油为购买各种各样的填充柱而四处忙碌和打听,最后还要花费相当多的银子去尝试不知道合适不合适的柱子,我替大家非常着急呀。因为好多方法和标准里面标注的色谱柱的填充和配制方法在实际中产生的效果不一定非常理想,因此大家为什么不自己尝试自己“制造”自己想要的宝贝呢?当然如果有的板油有其他的想法和潜规则俺就不多说拉。为了许多新板油的工作方便我还是从头到尾将色谱柱的:制造过程详细更大家交流以下,希望老专家和老板油为我的作法提出意见和建议,让我们的宝贝柱子更加听话。(听话我觉得用来形容色谱柱的性能是最合适不过的拉,因为我们的目的就是希望样品中各组分在色谱柱的作用下能按我们理想中的顺序和位置出现在谱图上,这就是我们对色谱柱最大的期望!)言归正传,开始陈述。一、所需要的设备和材料:1、真空泵:所需参数为抽气速率1极限压力≤ 6×10-2转速 1440电机功率 0.25进气口直径16泵油温升 ≤40用油量0.55价格一般在1500元左右,也就一两根柱子的价格,而且,它是色谱柱“制造”的最重要部件。http://ng1.17img.cn/bbsfiles/images/2011/10/201110110936_322873_1625930_3.jpg2、需要一个250mL或者500mL的玻璃压力缓冲瓶,上口连接真空泵,下口连接色谱柱。连接处使用密封带缠绕密封。缓冲瓶见下图http://ng1.17img.cn/bbsfiles/images/2011/10/201110111026_322887_1625930_3.jpg密封带如下图http://ng1.17img.cn/bbsfiles/images/2011/10/201110111030_322889_1625930_3.jpg3、空的色谱柱:空的柱子应该根据我们分析方法或者气相色谱仪的要求,有些气相色谱仪只能接2mm的柱子,有些只能接3mm的柱子。或者在柱子两端加上变头也可以兼容,但是那样比较麻烦呀!空的色谱柱可以使用新的,也可以使用失效的老色谱柱,但是都需要清洗一下,然后干燥。干燥的方法可以使用老化柱子的方法,非常好。下面我将我使用的一些柱子贡献出来让大家参观。呵呵http://ng1.17img.cn/bbsfiles/images/2011/10/201110111417_322939_1625930_3.jpghttp://ng1.17img.cn/bbsfiles/images/2011/10/201110111417_322940_1625930_3.jpghttp://ng1.17img.cn/bbsfiles/images/2011/10/201110111419_322942_1625930_3.jpg4、需要一些色谱柱的堵钭,我一般用细白钢丝。http://ng1.17img.cn/bbsfiles/images/2011/10/201110131416_323401_1625930_3.jpg将白钢网撕成宽0.5cm,长1cm左右的长条,卷成一卷,塞入色谱柱。二、色谱柱固定相的配制。 按照分析方法或者标准的要求用烧杯称取固定液和单体。使用合适的溶剂将固定液溶解,然后将单体倒入,轻轻晃动烧杯,一般不要使用搅拌棒,因为容易将单体搅碎拉!在使用红外线烘干!因为我没有红外线设备,所以使用常温(通风条件比较好)或者暖气哄干,效果还不错一般需要一周时间才能完全干燥。千万不要使用干燥箱,因为挥发的溶剂在干燥箱里聚集,容易发生爆炸事故!!!!关于称取单体和固定液的量应该根据自己的需要,一般我是每次配制固定相都是按一年的量计算,每次单体在50克左右。三、装柱等固定相完全干燥后就可以装柱拉。装柱的工作量最大,而且需要耐心,不要急噪。装柱的流程是真空泵,耐压管线(直径大小根据真空泵入口和缓冲瓶的口径决定),缓冲瓶,色谱柱连接检测器一端(使用白钢网封头),色谱柱连接汽化室一端,漏斗。然后使用密封带将各连接处缠绕密封好。接上电源,开启真空泵,检查是否漏气。如果漏气继续使用密封带密封,如果不漏气就可以装柱拉。等真空泵开启稳定1分钟后慢慢将固定相倒入漏斗,越慢越好,固定相在负压的作用下能够迅速到达色谱柱的检测器一端,使用金属工具慢慢敲打和震动色谱柱,使固定相能够填充密实,不要出现有空短的地方。使用这样的方法用固定相将色谱柱装慢,慢慢敲打震动,直到色谱柱的固定相不再下沉后才算柱子装好拉。最后停电源,拆卸刚才的密封连接,将色谱柱取下,从汽化室一端倒出一点点,使用白钢网堵上。最后一点最重要的是在色谱柱上挂上金属铭牌,写上色谱柱的一些参数和配制的日期。因为要是今后填充的柱子多拉容易搞混,如果忘记了这根柱子的参数,那这柱子就废拉,自己的工作也白费拉。四、试柱色谱柱装完后,将两端留在白刚网上的固定相倒出,不要带进色谱仪中。然后将色谱柱连接到汽化室上,另一端放空,使用氮气做载气将色谱柱老化8小时左右。当然有一些色谱柱老化时间更长!老化后将另一端接在合适的检测器上,使用合适的载气,设定好合适的参数,让仪器稳定下来后就可以试柱拉。试柱最主要的目的是考察色谱柱的分离度,如果不能到达我们期望的分离效果,通过调节流速看能不能达到。否则就得延长色谱柱的长度,调整固定液的比例,等等个,这些主要用在开发新的分析方法上。对于成熟的方法和技术,按要求做出来的色谱柱应该是八九不离十。如果色谱柱的分离很理想的话就可以使用拉。使用的使用如果是外标和相对归一法就得进标准样品,计算校正因子。如果是内标,就得往样品中加入内标物分析。好拉,自己说得很多拉。反正我知道自己“制造”色谱柱的好多益处,除了节约银子,工作方便,而且还能按照自己所需开发一些新的分析方法,自己“制造”色谱柱的好多益处,除了节约银子,工作方便,而且还能按照自己所需调节填充柱的一些参数,比如固定液的比例,柱子的长度等等来达到更好的分离效果。当然自己“制造”色谱柱也有一些不足,比如增加自己的工作量,不具备填充色谱柱的条件需要购买一些设备和试剂和耗材等。







 我要推广仪器
我要推广仪器
 下载APP
下载APP