TCD检测微量氢气时,用什么载气比较合适
请问对于氢气中微量氯化氢气体的分析,是否可用电子捕获检测器进行检测?若能用电子捕获检测器则该选择那种色谱柱?V(HCL)=10PPM
检测甲烷中的微量硫化氢和二氧化硫,能否用热导检测器,用什么色谱柱?
[img=,690,392]https://ng1.17img.cn/bbsfiles/images/2024/02/202402181722256130_1933_5510889_3.jpg!w690x392.jpg[/img]PDHID检测器,分析微量氢气、氧气、氮气。换完载气之后,在氧峰之前基线下降,出完氧峰之后的基线又正常了。请教基线下降的原因。
GB8981-88气体中微量氢气的测定-[url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]法(此方法选用气敏半导体作为色谱检测器来分析氮气、氧气、空气中微量氢气)求助:有否适用此分析方法的成型仪器? (或[url=https://insevent.instrument.com.cn/t/Mp]气相色谱仪[/url]、或其它仪器)如有相关仪器或资料请发送至 qiaozh@wepec.com

气相色谱fid检测器点火后忘记把氢气调小结果基线出现很多毛刺,,求解决http://ng1.17img.cn/bbsfiles/images/2017/10/2015012817000216_01_2736114_3.jpg
氢气中的硫化氢需要做定量检测,应该用什么检测器,最好能做到1ppm左右。进行简单前处理也可以。先谢过各位老师了
打算用气相色谱仪检测微量、常量氢化氰,常量氢化氰用TCD检测器,是否可以?微量氢化氰用NPD检测器,是否可以?请指教,谢谢!注:不想用化学法检测
GC112A色谱在使用载气为氢气,纯度99.9%,工业生产用,请问发生检测器堵塞是否因为氢气不纯造成的?我们用102G色谱时,也是用同一气体,流速相同,不同的地方是检测器与柱子同一温度,都在层析室内,温度不超过140度,请问PEGA是不是超过150度时,柱子固定相流失严重吗?请老师指导.多谢
ECD检测器污染,用氢气老化的色谱条件是什么?用氢气老化应注意些什么?
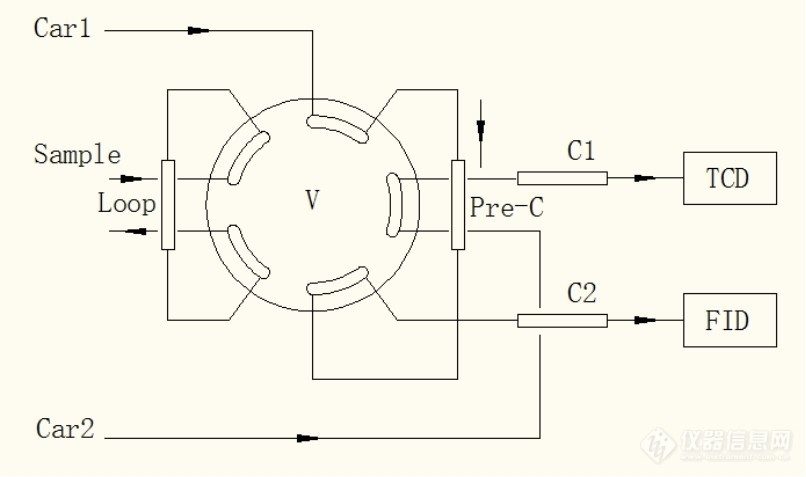
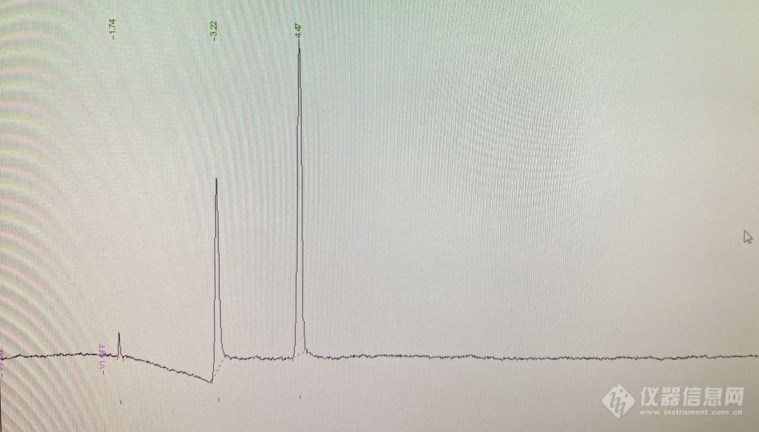
[align=center][size=24px]十通阀进样反吹分析化工排放空气中的微量氢气、氧气、丙烯腈[/size][/align][align=center]概述:[/align]采用常见的十通进样反吹方法,分析某化工企业工艺废气中的微量氢气、氧气和微量丙烯腈。[align=center]背景介绍[/align]某化工企业的分析要求:某合成工段排放废气(含量样品基质为空气,进样前处于常温常压状态,个目标组分含量大约数百ppm左右)中的氢气、氧气和微量的丙烯腈。[align=center]分析系统介绍[/align]于是大致设计了一下方案:首先选用Shimadzu公司的GC-2014C[url=https://insevent.instrument.com.cn/t/Mp]气相色谱仪[/url],安装有TCD检测器,以氩气作为载气,可以定量样品中的氢气、氧气和氮气(浓度范围为数百ppm左右)。色谱仪另外安装有FID检测器,用以分析样品中的微量丙烯腈(浓度范围为10ppm左右)。其次为色谱柱选择:为实现分离氢气、氧气、氮气的目的,一般会选用分子筛色谱柱。丙烯腈(样品中可能还有其他杂质,如水)的分离采用了有机担体固定相。单独测试有机担体色谱柱,丙烯腈色谱峰形较为理想,理论塔板数和不对称因子实验效果较好。阀系统的设计:鉴于分子筛柱会吸附微量丙烯腈,长时间使用会造成色谱柱分离性能下降,于是采用了经典的十通阀进样反吹的方案。样品在预柱上分离进行预分离,将氢氧氮和丙烯腈分离成为两组。预柱后面串联分子筛色谱柱,将氢气、氧气和氮气进行色谱分离。然后反吹预柱,将丙烯腈等其他杂质反吹到有机担体柱上,进行分离。进样步骤解析:1 下图为系统待机状态,在常见的进样反吹系统出口连接了有机担体柱。预柱中载气流向如图所示。此时,将样品气装载到定量环(Loop)中。[img=,690,408]https://ng1.17img.cn/bbsfiles/images/2021/10/202110052325593419_8983_1604036_3.jpg!w690x408.jpg[/img]2 进样十通阀旋转,系统状态如图所示:[img=,690,403]https://ng1.17img.cn/bbsfiles/images/2021/10/202110052326124515_4764_1604036_3.jpg!w690x403.jpg[/img]C1柱为分子筛色谱柱,气体流向如图中所示。氢气、氧气、氮气在TCD上依次出峰。3 反吹当氢气、氧气、氮气完全进入C1柱后,阀再次旋转,恢复到待机状态,此时预柱Pre-C载气反吹,样品中的丙烯腈在FID上出峰。[align=center]系统谱图:[/align][img=,443,424]https://ng1.17img.cn/bbsfiles/images/2021/10/202110052326275630_9806_1604036_3.jpg!w443x424.jpg[/img][img=,448,332]https://ng1.17img.cn/bbsfiles/images/2021/10/202110052326327383_7922_1604036_3.jpg!w448x332.jpg[/img][align=center][/align]
最近实验室需要购入一台[url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url],进样方式为手动注射器抽取气体样品注入,测定混合气体中的微量氢气含量(光催化产氢)请问哪家厂家的[url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]比较值得推荐另外请问,购买色谱并不附带色谱工作站吧,单独购买色谱工作站是不是智达的就可以?
我现在需要检测混合气体中微量氢气,组分可能有N[sub]2[/sub],O[sub]2[/sub],CO[sub]2[/sub],H[sub]2[/sub]O,现有[url=https://insevent.instrument.com.cn/t/Mp]气相色谱仪[/url]一台,配有FID和ECD检测器,还可以配个TCD检测器吗?谁那里有检测的方法啊,能告诉我不?
目前使用FID检测器检测,因为甲烷含量比较低含量1ppm,基线噪声干扰比较大,有没有什么新的方法或者灵敏度高一些的色谱推荐一下?
[em07] 我没有用过色谱分析微量的一氧化碳和二氧化碳,请问用哪一种仪器比较合适?是测量氢气中的微量CO和CO2的.
用什么型号的色谱可以测定 氢气中的微量CO和CO2?
[b][url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]氮磷检测器的使用与维护[/b]氮磷检测器(NPD)是分析微量含氮有机污染物的常用手段。关于NPD使用与维护的文献不多,不同厂家的NPD结构性能也有差异,本文以岛津色谱仪的氮磷检测器为例,依据作者使用经验,分析介绍NPD使用维护的一些问题。1 NPD的铷珠1.1 铷珠老化老化过程也是铷珠损耗过程,尽量减少高温阶段老化的时间,可以减少损耗。(1)在大部分的时间内,保持较低的老化温度和载气流量,当基线相对较为平稳时,再逐步升高温度,直至达到分析所需的温度和载气流量。(2)从老化温度接近分析所需温度时起,间断注入一些高浓度样品,有利于基线迅速平稳,缩短老化时间。1.2 铷珠加热电流调节氢气流量和铷珠加热电流,可以改变NPD的灵敏度,同时也影响铷珠消耗量。提高加热电流,NPD检测限可以优于厂商给出的技术指标1~2个数量级,但这是以铷珠寿命的损失为代价的,非必要不可如此。加热电流上限:从检测器上面的出气孔看到铷珠由暗红转为全部发红,就达到上限了。如果继续升高加热电流,虽然灵敏度能提高,但会大大增加铷珠消耗速度。从节省的角度,当使用NPD时,调节加热电流使灵敏度达到基本的分析要求就可以了,不必太高。1.3 固定相的影响除了含氰固定液伤害铷珠,不可使用外,硅氧烷固定相流失会污染检测器,也不宜使用。在微量组分分析时,常对进样口的玻璃衬管、玻璃柱和石英棉进行硅烷化处理,以减少吸附,提高分离效果。但使用NPD时建议不要这样做,因为流失的硅烷化试剂会污染铷珠,降低NPD的灵敏度。有文献报道,色谱柱和石英棉用H3PO4处理,有利于NPD提高灵敏度和稳定性。2 色谱分析系统2.1 气路系统的稳定性除了对气体纯度的要求之外,在高灵敏度分析时,NPD对气体流量的稳定性有更高的要求。特别要注意的一点是,有些型号的岛津色谱FID是双系统(双气化室!双检测器)结构,供气方式是两系统共用一个流量计并联供气,NPD安装在其中一个FID的基座上,使用其中一组系统。这样,另一闲置的气化室和FID检测器就成为气路系统的漏洞。解决气化室的问题很简单,将其上下端用硅胶塞和封口螺丝堵上就可以了 而闲置的FID检测器漏气的问题因与对仪器内部气路系统的了解与熟悉程度有关,常常不被注意,以致于仪器始终达不到高的灵敏度和稳定性。解决的方法也比较麻烦,要拆开仪器,将通往该检测口的气路堵死。这一点对于NPD和FID经常要互换的仪器来说,很麻烦,但很重要。2.2 分析系统的洁净
[em0716] 现在有的条件是 6890 [url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url],载气为氮气,多种组分的气体为:氢气、甲烷、氮气、氧气、一氧化碳、二氧化碳。请教问题: 1、气体如何收集? 待检测的气体流量不是很大。现有的收集设备是10ml Aglient的进样针,不知取样后待测气体是否会被空气所污染? 2、色谱需要设定怎样的条件? 做了几个样,由于用氮气作为载气,检测出的氢气峰是倒峰,而且出现的峰主要只有一个,估计是氢气,几个样的峰尖位置时间都不在同一点,不知道什么原因造成? 3、如何通过出现的峰来反应出氢气的含量? 从实验条件考虑,用外表法得到特征曲线(具体用空气稀释纯氢得到不同含量的氢气标样),此方法是否合适?是否适用此种多组分的气体?或者是否有更好的方法? 谢谢帮忙解决@!!
最近要用到[url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]的FID检测器,但是没有氢气,老师说可以用氢气发生器产生,但是我怀疑纯度,而且不知道怎么将氢气发生器和色谱联系起来,求高手指教啊!
请问专家怎样用氢气为载体,以[url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]测量微量水?
2月份新买安装的设备。配了TCD/FID检测器。安装时,工程师帮忙安装了HP-5的色谱柱到TCD上(FID也连接并测试正常后换到TCD),因最近才要用到FID,自己安装好HP1柱子。使用时才发现FID的氢气流量为负值。在主机键盘上找到FID的氢气流量选项,调为ON的,显示也为打开。但流量还是为负值。打电话到AGlilent,答复要1个星期才派人来修。算算这才第二次使用到FID,就出现这种问题!有点搞笑。。不知哪位大大了解这种情况的,帮忙解答下。
色谱的色谱柱活化后开始降温时,能否先关闭氢气和空气让检测器降温快点?
各位大神,国产机器GC 2000 III仪器,TCD检测器,氢气做载气,色谱柱老化应该怎么办,需要换载气吗?平时工作状态是,柱箱70度,进样口80度,检测器100度。色谱柱是填充柱,比较粗的那种,老化的时候能不能把温度调高走几个小时就行?
[align=center][b][size=18px]气相色谱仪FID检测器结构特点、基本操作、常见故障及排除[/size][/b][/align] 在气相色谱仪众多检测器中,FID检测器(氢火焰离子化检测器)是气相色谱蕞常用一种检测器,它具有灵敏度高、线性范围宽、应用范围广、易于掌握等特点,特别适合于毛细管气相色谱。FID检测器对大多数有机化合物有很高的灵敏度,灵敏度比热导检测器TCD高100-10000倍。[b]一、结构特点[/b] 气相色谱仪FID检测器由离子座、离子头、极化线圈、收集极、气体供应等部分组成,离子头是检测器的关键部分。 微量有机组分被载气带入检测器以后,在氢火焰的作用下离子化。产生的离子在发射极和收集极的外电场作用下定向运动形成微电流。有机物在氢火焰中离子化效率极低,估计每50万个碳原子仅产生一对离子。离子化产生的离子数目,在一定范围内与单位时间进入检测器的被测组分的质量成正比。 微弱的离子电流经高电阻(108~1011 Ω)变换成电压信号,经放大器放大后,由终端信号采集即得出色谱流出曲线。在正常点火的情况下FID信号大小受离子化效应和收集效应的影响。其中离子化效应的影响因素有样品性质(不同的物质校正因子不同)和火焰温度(受几种气体的流量比影响);收集效应的影响因素有极化电压和喷嘴、极化极、收集极的相对位置。因此对同一样品要获得高灵敏度必须选择蕞佳氢气、载气、空气的流量比;蕞佳的喷嘴、极化极、收集极的相对位置与适当的极化电压。氢气、载气、空气的流量可通过实验摸索蕞佳条件,一般理论比为30∶30∶300。[b]二、基本操作[/b] 1)拧开各气体总压开关(逆时针旋转为开) ,旋转各调节阀,使各压力表 指示在 0.3~0.4 MPa(顺时针旋转为开) 。 2) 通入载气 2) 将载气流量调至 20~30ml/min (N , (载气压力表 1: 0.05MPa; 。 载气压力表 2:0.03 MPa) 3) 通载气约 10min 后 (若长期停机后重新启动操作时, 通载气 15min 以上) , 开启色谱仪电源总开关,设置所需柱箱、汽化、检测器 2 的工作温度。 柱箱温度必须低于色谱柱固定相蕞高使用温度(不锈钢色谱柱的使用温 度≤230℃, 毛细管色谱柱的使用温度≤300℃) 汽化室和检测器温度必须 , 高于 100℃(若无高沸点的组分一般设置 150℃) ,设置好后按运行键即 可升温。 4)将“灵敏度选择”置于 2 档,讯号衰减开关置于 1 档。打开微电流放大器 开关,旋转零位调节电位器,使基线在零位附近(在此之前应打开计算 机,进入 1 通道界面) 。 5)旋转空气流量调节阀,将空气流量调至 200~300 MPa(空气压力表指示 在 0.02~0.03 MPa,一般调至 0.03 MPa)待检测器温度升到 100℃时,即 可打开 H2,并旋转 H2 调节阀到压力表指示 0.02 MPa 附近,打开 H2 点 火开关阀,用电子点火枪在 FID 检测器出口处点火,点燃后关闭 H2 点 火开关阀。 6)待基流稳定后,准备进样(一般进样量为 0.4~0.5ml),进样后立即按下 带有“A”字样的按扭,此时开始采样。 7)当所有测试完毕停机时,必须先将 H2 开关阀关闭,再将微电流放大器 开关关闭,退出升温开始降温,待柱箱温度降至室温,汽化和检测器温 度降至 70℃以下时,关闭载气、空气、H2 和色谱仪电源总开关。[b]三、常见故障及排除[/b] 1、 进样后色谱不出峰 故障原因及排除方法如下: (1)未点着火 首先用一冷的光亮的铁板置于检测器的上方,若有细小水珠生成,则证明火已点着;反之证明火未点着,此时,需检查氢气、氮气、空气的密封情况是否完好,是否有漏气现象。其次用皂沫流量计测量流速是否正常,适当增大氢气的流速,减小载气与空气的流速,待点着火后再将各流速调至蕞佳流速位置。 (2)信号输出中断 检查从色谱仪到工作站的信号线连接情况,观察有无接触不良或断开的情况。另外,在进样后用万用表测量色谱信号输出,观察有无信号输出,若无信号输出则证明此故障由色谱仪引起,需做进一步检查。 (3)收集极绝缘不好 测量收集极与仪器外壳的电阻应大于1013 Ω。 (4)其它方面的原因 主要包括进样垫损坏、色谱柱断裂(毛细管柱比较常见)、微量进样器损坏等。 2、基线噪声波动大 (1)电器方面的原因 首先将检测器信号线断开,在采集状态下观察基线运行情况,如果基线波动很大则可判断该故障是电器方面的原因,此时,需要进一步检查仪器接地是否良好(接地电阻应小于5 Ω)、线路板及各插件是否松动等。 (2)测量系统污染 断开信号线后,在采集状态下检查基线运行的情况,如果基线运行正常则证明测量系统污染。需要检查色谱柱是否失效(需活化处理)、柱进口是否污染(更换玻璃丝、玻璃衬管等)、检测器污染,主要是离子头的污染,因为此处高温会有杂质碳结,需要小心拆下检测器用中性溶剂清洗。 3、空气峰掩盖组分峰 分析微量组分时,如分析液态氧气中总烃含量时,氧信号峰保留时间蕞小,随后是甲烷、乙烷、乙烯等,如果调整不好会出现氧气覆盖甲烷或将氧气峰误判为甲烷峰。排除办法是逐渐降低氢气流速,依次进样可观察到氧气峰逐渐降低,调节至满意为止。

有一段时间没有用FID检测器了,今天开起来使用时发现FID检测器氢气流量无法上去约0.3ml/min(设定值30ml/min)查看氢气瓶还剩5mpa气体,进样口垫片刚更换进样口压力正常,不知道是不是色谱柱至检测器之间哪里漏气的原因,要如何确认。望得到有处理过此类问题的版友帮助,谢谢问题找出来了,是压力表减压阀被关小了,导致压力不足所致,减压阀重新调整到原先位置,仪器就可以正常使用了http://ng1.17img.cn/bbsfiles/images/2012/04/201204121046_360837_2357013_3.jpg
刚开机或关机重启时,氢气流量能达到设定值,90ml/min,但点火几秒后就息了。氢气、空气缓慢降至0 。.关闭点火开关,重新打开氢气空气,空气能达到设定值,但氢气只能在八十多就上不去了,也点不燃火。[img]https://simg.instrument.com.cn/bbs/images/default/em09509.gif[/img]使用的是氢气发生器,氢气发生器正常,不连接[url=https://insevent.instrument.com.cn/t/Mp]气相色谱仪[/url]能达到最大流量。色谱柱连接检测器一端,取出来看了一下,长度超出了规定长度一些,切掉重新量了长度重装了柱子,依旧是上面的情况。FPD检测器自从买回来,就每次检定的时候才用过,完全看不明白是怎么回事啊,走过路过的同志们帮忙看看是怎么了。前两天使用FID检测器的时候,是能正常点火使用的,但FID检测器用的氢气流量没这么大,只有40.
用什么样的柱子\检测器利用[url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]法分析苯胺中的微量硝基苯(0.5ppm以下)
氢气发生器的氢气会不会携带微量的碱蒸汽?
我单位新购了一台配置PFPD检测器的瓦里安CP3800[url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url],用氢气发生器提供H2,在样品较多时,能否无人值守24小时工作?会不会有什么危险?
谢谢专家指导!条件如下:用GC112A色谱,氢气作载气,柱前压1.2公斤,汽化温度185度,柱温130度,检测150度TCD检测器,桥流180,固定相10%PEGA,担体为401,60-80目,柱子为内径4mm,长2米.用于分析醋酸乙酯成品及粗品.粗品中成分大致如下:水 7-10% 酒精 2-4% 醋酸甲酯 2-4%,其余为醋酸乙酯。成品中水小于0.2%,酒精小于0.2%,其余为醋酸乙酯。问题是:水与酒精分离不好,水严重拖尾。







 我要推广仪器
我要推广仪器
 下载APP
下载APP