珀金埃尔默Torion助力新国标《水中挥发性有机物的测定便携式顶空/气相色谱质谱法》
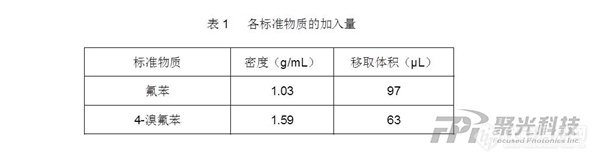
近期,生态环境部办公厅发布了《水质挥发性有机物的测定 便携式顶空/气相色谱质谱法(征求意见稿)》,该标准规定了地表水、地下水、生活污水、工业废水和海水中挥发性有机物的现场快速定性和56种目标化合物的定量分析。珀金埃尔默Torion T-9仅需80秒即可完成标准中56种VOCs的定性定量分析,可从容应对环境突发事件的应急监测需求。减少了样品运输和保存过程中待测物质的变化,具有实验室分析方法不可替代的优势。随着我国经济的增长,工业发展迅猛,在化工品生产、运输和储存过程中导致的挥发性有机物(VOCs)污染事故频发,严重影响了当地的人民生活、社会稳定和经济发展。VOCs并非单一的化合物种类众多,具有迁移性、持久性和毒性是一类重要的环境污染物。VOCs会对空气、水、土壤等造成严重伤害和污染,其中水与我们的生活息息相关。目前,国内外针对水中VOCs的检测标准主要是顶空气相色谱法、顶空气相色谱质谱法、吹扫捕集气相色谱质谱法等均为实验室检测标准。珀金埃尔默Torion T-9便携式气质配合SPS-3顶空工作站可以在突发应急现场分析水中VOCs,样品分析速度快,检测56种VOCs仅需80秒,同时峰形尖锐分离效果好。在满足新标准的同时可在突发性环境应急事件中快速提供检测结果,指导应急策略。Torion T-9便携式气质技术优势:SPME/CME/顶空/热脱附等多种样品前处理方式创新的环状离子阱比常规离子阱离子容量高400倍开机5分钟做样3分钟升温速率高达2.5℃/s无基础用户一天培训可独立操作隔膜泵/涡轮分子泵的真空系统非耗材省心省成本图1 56种VOCs与2种内标总离子流图1-氯乙烯;2-1,1-二氯乙烯;3-二氯甲烷;4-反-1,2-二氯乙烯;5-1,1-二氯乙烷;6-氯丁二烯;7-顺-1,2-二氯乙烯;8-2,2-二氯丙烷;9-溴氯甲烷;10-氯仿;11-1,1,1-三氯乙烷;12-1,2-二氯乙烷;13-1,1-二氯丙烯;14-苯;15-四氯化碳;16-1,2-二氯丙烷;IS1-氟苯(内标);17-三氯乙烯;18-二溴甲烷;19-一溴二氯甲烷;20-顺-1,3-二氯丙烯;21-反-1,3-二氯丙烯;22-1,1,2-三氯乙烷;23-甲苯;24-1,3-二氯丙烷;25-二溴氯甲烷;26-1,2-二溴乙烷;27-四氯乙烯;28-氯苯;29-1,1,1,2-四氯乙烷;30-乙苯;31/32-对/间-二甲苯;33-溴仿;34-苯乙烯;35-邻-二甲苯;36-1,1,2,2-四氯乙烷;37-1,2,3-三氯丙烷;38-异丙苯;39-溴苯;40-正丙苯;41-2-氯甲苯;42-4-氯甲苯;43-1,3,5-三甲基苯;44-叔丁基苯;45-1,2,4-三甲基苯;46-1,4-二氯苯;IS2-1,4-二氯苯-d4(内标);47-仲丁基苯;48-1,3-二氯苯;49-4-异丙基甲苯;50-1,2-二氯苯;51-正丁基苯;52-1,2-二溴-3-氯丙烷;53-1,2,4-三氯苯;54-萘;55-六氯丁二烯;56-1,2,3-三氯苯;图2 1,2-二氯丙烷、三氯乙烯、二溴甲烷和一溴二氯甲烷共流出解卷积谱图在突发应急事件中,由于便携质谱检测结果是制定应急决策的重要依据,不但要快而且要准。Torion T-9内置强大的谱库的同时还具备独特的解卷积功能,可以轻松鉴定极为复杂的化合物,即使有化合物共流出也可以实现准确定性和定量。如图2所示1,2-二氯丙烷、三氯乙烯、二溴甲烷和一溴二氯甲烷共流出通过Torion T-9的内置谱库和解卷积功能可以准确识别出这4种物质。Torion T-9便携式气质为突发应急保障而设计,总重量仅14.5公斤,仪器从启动到样品分析仅需5分钟,样品分析时间3分钟以内,在福建泉港C9泄露、江苏海安工业园泄露、青岛上合峰会、武汉军运会等突发事件和重大会议保障上起到了关键的作用。



























 我要推广仪器
我要推广仪器
 下载APP
下载APP