我之前定量没有用同位素内标,保留时间也能分开。最近看到关于质谱定量内标选择的内容,认为同位素内标好,能校正基质效应,色谱行为和响应特征接近,即便没有合适的同位素内标,也要选择结构类似的,色谱行为接近的。但是如果液相分不开一起进质谱的话,会不会产生离子抑制?
你好,如果我采用质谱定量,用内标法,内标的含量保持恒定,按比例增加标准样品的浓度,以标准样品和内标的比值及标准样品的浓度做标准曲线,会不会因为标样的浓度越来越大,产生离子抑制,而使标准曲线趋势降低,结果偏低?
用质谱测定血清中的药物浓度,需要加入20ul的内标,但内标不溶于水,溶于甲醇,所以内标加进去后可能会有蛋白沉淀,这样会不会导致内标分布不均匀?并且,离心之后还是存在这样的现象。这样的现象主要是在同一个样品连续进样后,发现内标的响应相差悬殊才发现的。各位大侠有没有发现类似的问题?
[font=宋体][size=10.5pt]HJ 805-2016 [font=宋体]土壤和沉积物 多环芳烃的测定 [url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url][/font][font=Times New Roman]-[/font][font=宋体]质谱法。分析定量时,标准曲线的绘制时,标准上说以目标化合物和内标物浓度的比值为横坐标;以目标化合物定量离子响应值和内标化合物定量离子响应值的比值与内标化合物质量浓度的乘积为纵坐标。绘制标准曲线。这样是不是就不能像外标法一样,仪器的数据处理软边可以直接给出标曲,需要自己手动算了?谢谢各位![/font][/size][/font][font=宋体][size=10.5pt]还有就是如果按照以上说的方法绘制曲线,十几种目标物只有5种内标物,没有相对应的内标物的目标化合物应该怎么绘制标准曲线?[/size][/font][font=宋体][size=10.5pt]内标法实在是小白一个?希望大家多多解惑,非常感谢[/size][/font]
质谱:OmniStar/ThermoStar软件:Quadera 4.4问题:想用He做内标在线定量分析反应,没有连色谱,所有的产物都是气体实验室以前没有人用质谱,这台质谱老师只会定性分析MID模式,从来没有做过定量,现在完全不知道要如何设置和操作,求各位大神帮忙
质谱:OmniStar/ThermoStar软件:Quadera 4.4问题:想用He做内标在线定量分析反应,没有连色谱,是直接反应气连到质谱的实验室以前没有人用质谱,这台质谱老师只会定性分析MID模式,从来没有做过定量,现在完全不知道要如何设置和操作,求各位大神帮忙
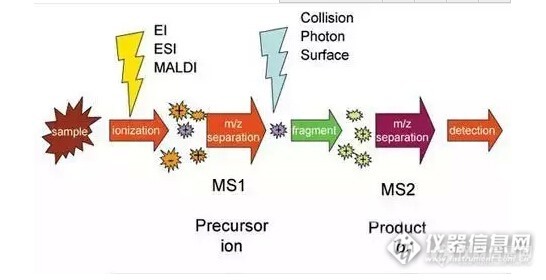
质谱信号。与EI谱图分析以相对强度为主不同,在色谱-质谱联用时,信号的绝对强度就成了我们天天都要关心的内容,因为质谱信号强度随时间的变化就是实验的色谱图,通常以总离子强度或者某一特定质荷比离子的强度作图。http://ng1.17img.cn/bbsfiles/images/2015/11/201511271813_575350_2544766_3.jpg2、定量的两种方法外标法 用已知量的标准样品A和未知量的待测样品A分别进行实验;我们会得到以下三个信息:标准样品的量(已知);标准样品的信号强度;待测样品的信号强度。(假设样品的响应=常数*浓度,从这三个信息即可算出待测样品的量。) 为了更加精确地测定未知量的样品,我们希望标准样品的信号强度与待测样品的信号强度尽量接近(以减少非线性响应的影响)。因此常用的外标法会测量一系列已知量的标准样品,绘制一条工作曲线,再用拟合的方法确定未知样的量。http://ng1.17img.cn/bbsfiles/images/2015/11/201511271814_575351_2544766_3.jpg内标法 外标法主要有以下两方面的局限:1标样和待测样是独立进行实验的,实验间的偶然误差无法消除;2标样和待测样的基质(即除待分析物外的其它成分)不同,基质有可能会带来不同的影响,也会产生误差。 那么,如果我们把已知量的标准样品B直接加入待测样品A,就可以把标准样品和未知样品的测定在同一次实验和同样基质中完成,也就消除了两次实验和基质不同造成的误差,这就是内标法。(如果加入的标准样品和待测样品是同种物质A,那么由于它们不可区分,只通过一次实验是不能定量待测样的,这时我们在加入标样前后分别进行两次测量,即测量待测样及待测样+标样的信号,即可计算出待测样的量。)3、质谱相关的特殊定量细节同位素稀释 前面内标法的介绍中我们可以发现,最理想的内标物既要和待测样相同(具有相同的响应系数)又要不同(仪器可以区分二者的信号),这对矛盾的集合体就是同位素内标。 由于不同同位素的化合物具有近似相同的物理化学性质,离子化时的响应通常也是相同的,而它们具有不同的质荷比m/z,即可在质谱中被区分出来。因此同位素标准品是最理想的内标物。 另外,由于某些元素的天然同位素分布有一定的比例,当我们加入一定量的同位素内标时,可以把对信号绝对强度的测量转化为对信号相对比例的测量,从而提高实验的准确性。http://ng1.17img.cn/bbsfiles/images/2015/11/201511271814_575353_2544766_3.jpg选择反应监测 在不太复杂的体系中,我们只要按照分子量就可以定性某种化合物了。但对于复杂混合物(如石油产品/生物样品)而言,很多化合物具有相同或相近的质量(同分异构体质量完全相同,有些化合物分子量非常接近,如CO和N2,要考虑仪器的质量分辨率是否能区分二者),此时仅靠测量质量就不能确定这个化合物是否就是我们关心的“the one”了。 在串联质谱 (Tandem MS) 仪器中,我们不仅可以把质谱仪理解为一个称量离子的“天平”,它还具有了离子“镊子”(选择某个特定的离子把它分离出来)和“剪刀”(把某个/某些离子激活并打成碎片)的功能。通过母离子和子离子的两步选择,我们可以在复杂体系中精确定位到我们关心的化合物,同时,两次离子选择还可减少复杂基质的干扰,降低背景噪声(获得更低的检出限)并提高方法的动态范围。因此选择反应监测是目前色谱(气相色谱/液相色谱)-质谱联用中最常用的定量方法。http://ng1.17img.cn/bbsfiles/images/2015/11/201511271815_575354_2544766_3.jpg选择反应监测在不太复杂的体系中,我们只要按照分子量就可以定性某种化合物了。但对于复杂混合物(如石油产品/生物样品)而言,很多化合物具有相同或相近的质量(同分异构体质量完全相同,有些化合物分子量非常接近,如CO和N2,要考虑仪器的质量分辨率是否能区分二者),此时仅靠测量质量就不能确定这个化合物是否就是我们关心的“the one”了。在串联质谱 (Tandem MS) 仪器中,我们不仅可以把质谱仪理解为一个称量离子的“天平”,它还具有了离子“镊子”(选择某个特定的离子把它分离出来)和“剪刀”(把某个/某些离子激活并打成碎片)的功能。通过母离子和子离子的两步选择,我们可以在复杂体系中精确定位到我们关心的化合物,同时,两次离子选择还可减少复杂基质的干扰,降低背景噪声(获得更低的检出限)并提高方法的动态范围。因此选择反应监测是目前色谱(气相色谱/液相色谱)-质谱联用中最常用的定量方法。
土壤国标现在大部分是用质谱做的都是内标法,为什么不用外标,感觉内标法好麻烦!!!!
色谱定量中,内标法和内标百分比法的区别是什么?归一化法和校正面积归一化法的区别又是什么?求详解。
色谱定量中内标法与外标法各有什么优势?
色谱定量分析中 选内标法或外标法 ?
[color=#444444]最近在做瘦肉精及降糖中药中化学添加成分的测定,使用液相-离子阱质谱来分离测定,可是发现一般国标及文献上都用的是氘代内标来定量,经费不足啊!那些内标都好贵啊!单纯用外标法来定量的话行不?有没有什么好的办法?另外有没有虫友用过天津光复的色谱纯试剂,用国产的色谱纯试剂做流动相行不行?[/color]
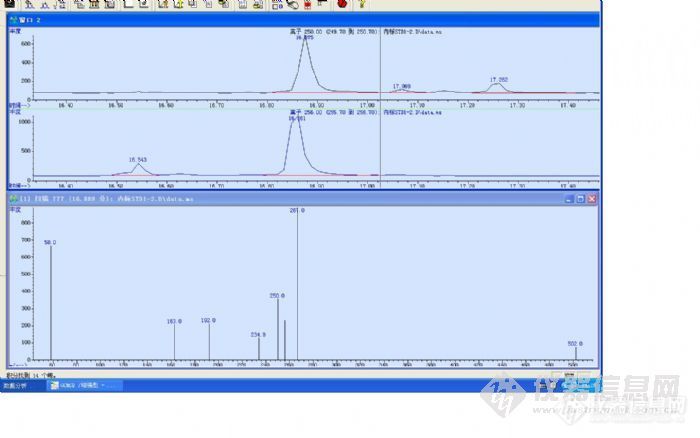
本人对气质的内标定量法存在一个问题,现发帖向各位大侠求解,希望大家踊跃探讨!!问题:A物质的定量离子是250B物质的定量离子是256B物质是A物质的内标同位素出峰的时候A和B是没有分开的http://ng1.17img.cn/bbsfiles/images/2011/09/201109261440_319374_1967303_3.jpg从质谱图中可以看出,两个峰是重叠的,就是说A物质中有B物质的定量离子256,B物质中又有A物质的定量离子250,这样的话,同位素B的250离子会不会增加了A物质原来的含量呢?A物质又会不会增加了B的实际含量呢?这样对检测结果的含量会不会有所偏大呢??
各位老师、大侠们, 今天收到审稿意见,即:“与外标法相比,内标法的优点可否定量体现”,我们的试验是采用内标法建立标准曲线定量分析计算,请问我该如何回答审稿意见呢?翻阅 汪正范编著的《色谱定性与定量》,其中说道“内标法定量比标准曲线法定量的准确度和精密度都要好”
三重四级杆质谱是如何定量的?三重四级杆通过离子打碎获得特异性子离子,子离子在通过Q3后时在接收器上转化为电信号,反映到分析软件就是离子强度图。在一定线性范围下,分析物浓度越高,打到接收器上的离子就越多,信号越强,这是定量的基础。此外在定量时可以选择用峰高或峰面积定量,一般选用峰面积定量准确。因为是定量,所以必需标准曲线,原理与高效[url=https://insevent.instrument.com.cn/t/5p][color=#3333ff]液相[/color][/url]类似。一般多用内标法定量,以排除质谱重现性和样品处理造成的影响。那么,随着设置离子对越多,如何能保证定量准确呢?四极杆对于离子的选择性通过是交替进行的,在所有离子通道之间快速切换。随着设置离子对数量的增加,每个离子所占用的检测时间缩短,这会影响到其检测灵敏度,但是只要实际样品和标准溶液所采用的分析方法一致,定量的准确性理论上是不受影响的。
水质 松节油的测定 吹扫捕集 [url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]-质谱法 HJ 866-2017上述标准中,松节油混标里面的a-派烯、b-派烯要用内标法定量然而,松节油标液只有总的松节油浓度,没有给出a-派烯、b-派烯的浓度比例关系,怎么实现内标法定量?用的是岛津的GCMS
色谱分析的重要作用之一是对样品定量。而色谱法定量的依据是:组分的重量或在载气中的浓度与检测器的响应信号成正比。常见定量分析方法分为面积归一化法、内标法、外标法、标准曲线法等。大家常常容易傻傻分不清楚的莫过于内标法、外标法了http://simg.instrument.com.cn/bbs/images/default/em09512.gif以内标法为例,选一与欲测组分相近但能完全分离的组分做内标物(当然是样品中没有的组分),然后配制欲测组分和内标物的混合标准溶液,进样得相对校正因子。再将内标物加入欲测组分的样品中,进样后测得欲测组分和内标物的定量参数,用内标法公式计算即可。其实,从定义上来区分的话,外标法就是用标准品的峰面积或峰高与其对应的浓度做一条标准曲线,测出样品的峰面积或峰高,在标准曲线上查出其对应的浓度,这是最常用的一种定量方法;内标法是对应外标法说的,内标法是将一定量的纯物质作内标物,加入到准确称量的试样中,根据被测试样和内标物的质量比及其相应的色谱峰面积之比,来计算被测组分的含量。外标法需要用样品和标准品对比,但是有时我们很难保证样品和标准品进的体积是一样的,毕竟要有误差,这时候就用内标法,就是在外标法的基础上,在样品和标准品里在加入一种物质,通过加入物质的峰面积或峰高的变化,就可以看出我们标准品和样品进样体积的差别,但同时会引进加入物质的秤量误差。所以一般用外标法来定量,如果进样体积很难掌握,就用内标法,可以消除进样体积的误差。标准加入法标准加入法
[font=&]本期视频介绍一下关于内标法定量的基础知识,很多实验中,外标法定量会不准确,此时就需要用到内标法了。[/font]
请问什么是同位素稀释气相色谱质谱法?和同位素内标法一样吗?
大家好,本人是刚接触安捷伦气相色谱质谱6890A/5975c的同学,想用它来检测细胞内代谢物,选用了十四酸做内标,但细胞内代谢物各组分不知道,也无法得到标准品,请问这时候想用内标法定量细胞内的某些成分还能做么?如果能,具体怎样做才可以呢
内标法内标法是在试样中加入一已知量的标准元素s,其谱线的波长与分析元素A的分析线相近,通过测定它们的强度比以进行定量分析。因标准元素s需引入试样之内,故称之为内标元素,所选用的内标元素的谱线称为内标线,分析线与内标线合在一起称为分析线对,它们的强度分别以IA和Is表示,分析线对的强度比与基体无关,正比于分析元素的浓度比,即cA/cs=k IA/Is其中k为换算系数,可用已知分析元素和内标元素的标准样品事先求出。由于在此法中引入的元素s为已知量,因此在测出分析线对的强度后,可以用上式,或利用一组标样绘制的IA/Is-cA校准曲线,求出试样中分析元素的含量。 内标法可有效补偿元素间的吸收增强效应和长时间的仪器漂移,以及部分地补偿粉末和样品的密度变化,提高测定结果的准确度。
一、内标法什么叫内标法?怎样选择内标物? 内标法是一种间接或相对的校准方法。在分析测定样品中某组分含量时,加入一种内标物质以校谁和消除出于操作条件的波动而对分析结果产生的影响,以提高分析结果的准确度。 内标法在气相色谱定量分析中是一种重要的技术。使用内标法时,在样品中加入一定量的标准物质,它可被色谱拄所分离,又不受试样中其它组分峰的干扰,只要测定内标物和待测组分的峰面积与相对响应值,即可求出待测组分在样品中的百分含量。采用内标法定量时,内标物的选择是一项十分重要的工作。理想地说,内标物应当是一个能得到纯样的己知化合物,这样它能以准确、已知的量加到样品中去,它应当和被分析的样品组分有基本相同或尽可能一致的物理化学性质(如化学结构、极性、挥发度及在溶剂中的溶解度等)、色谱行为和响应特征,最好是被分析物质的一个同系物。当然,在色谱分析条什下,内标物必须能与样品中各组分充分分离。需要指出的是,在少数情况下,分析人员可能比较关心化台物在一个复杂过程中所得到的回收率,此时,他可以使用一种在这种过程中很容易被完全回收的化台物作内标,来测定感兴趣化合物的百分回收率,而不必遵循以上所说的选择原则。 在使用内标法定量时,有哪些因素会影响内标和被测组分的峰高或峰面积的比值? 影响内标和被测组分峰高或峰面积比值的因素主要有化学方面的、色谱方面的和仪器方面的三类。 由化学方面的原因产生的面积比的变化常常在分析重复样品时出现。 化学方面的因素包括: 1、内标物在样品里混合不好; 2、内标物和样品组分之间发生反应, 3、内标物纯度可变等。 对于一个比较成熟的方法来说,色谱方面的问题发生的可能性更大一些,色谱上常见的一些问题(如渗漏)对绝对面积的影响比较大,对面积比的影响则要小一些,但如果绝对面积的变化已大到足以使面积比发生显著变化的程度,那么一定有某个重要的色谱问题存在,比如进样量改变太大,样品组分浓度和内标浓度之间有很大的差别,检测器非线性等。进样量应足够小并保持不变,这样才不致于造成检测器和积分装置饱和。如果认为方法比较可靠,而色谱固看来也是正常的话,应着重检查积分装置和设置、斜率和峰宽定位。对积分装置发生怀疑的最有力的证据是:面积比可变,而峰高比保持相对恒定, 在制作内标标准曲线时应注意什么? 在用内标法做色话定量分析时,先配制一定重量比的被测组分和内标样品的混合物做色谱分析,测量峰面积,做重量比和面积比的关系曲线,此曲线即为标准曲线。在实际样品分析时所采用的色谱条件应尽可能与制作标准曲线时所用的条件一致,因此,在制作标准曲线时,不仅要注明色谱条件(如固定相、柱温、载气流速等),还应注明进样体积和内标物浓度。在制作内标标准曲线时,各点并不完全落在直线上,此时应求出面积比和重量比的比值与其平均位的标准偏差,在使用过程中应定期进行单点校正,若所得值与平均值的偏差小于2,曲线仍可使用,若大于2,则应重作曲线,如果曲线在铰短时期内即产生变动,则不宜使用内标法定量。 二、外标法 用待测组分的纯品作对照物质,以对照物质和样品中待测组分的响应信号相比较进行定量的方法称为外标法。此法可分为工作曲线法及外标一点法等。工作曲线法是用对照物质配制一系列浓度的对照品溶液确定工作曲线,求出斜率、截距。在完全相同的条件下,准确进样与对照品溶液相同体积的样品溶液,根据待测组分的信号,从标准曲线上查出其浓度,或用回归方程计算,工作曲线法也可以用外标二点法代替。通常截距应为零,若不等于零说明存在系统误差。工作曲线的截距为零时,可用外标一点法(直接比较法)定量。 外标一点法是用一种浓度的对照品溶液对比测定样品溶液中i组分的含量。将对照品溶液与样品溶液在相同条件下多次进样,测得峰面积的平均值,用下式计算样品中i组分的量: W=A(W)/(A) 式中W与A分别代表在样品溶液进样体积中所含i组分的重量及相应的峰面积。(W)及(A)分别代表在对照品溶液进样体积中含纯品i组分的重量及相应峰面积。外标法方法简便,不需用校正因子,不论样品中其他组分是否出峰,均可对待测组分定量。但此法的准确性受进样重复性和实验条件稳定性的影响。此外,为了降低外标一点法的实验误差,应尽量使配制的对照品溶液的浓度与样品中组分的浓度相近。 外标法 external standard method 色谱分析中的一种定量方法,它不是把标准物质加入到被测样品中,而是在与被测样品相同的色谱条件下单独测定,把得到的色谱峰面积与被测组分的色谱峰面积进行比较求得被测组分的含量。外标物与被测组分同为一种物质但要求它有一定的纯度,分析时外标物的浓度应与被测物浓度相接近,以利于定量分析的准确性。三、定量分析中怎样选择内标法或外标法 选一与欲测组分相近但能完全分离的组分做内标物(当然是样品中没有的组分),然后配制欲测组分和内标物的混合标准溶液,进样得相对校正因子。再将内标物加入欲测组分的样品中,进样后测得欲测组分和内标物的定量参数。用内标法公式计算即可。 内标法是将一定量的纯物质作内标物,加入到准确称量的试样中,根据被测试样和内标物的质量比及其相应的色谱峰面积之比,来计算被测组分的含量。 选择内标物有4个要求:1.内标物应是该试样中不存在的纯物质;2.它必须完全溶于试样中,并与试样中各组分的色谱峰能完全分离;3.加入内标物的量应接近于被测组分;4.色谱峰的位置应与被测组分的色谱峰的位置相近,或在几个被测组分色谱峰中间。 内标法的优点是测定的结果较为准确,由于通过测量内标物及被测组分的峰面积的相对值来进行计算的,因而在一定程度上消除了操作条件等的变化所引起的误差。内标法的缺点是操作程序较为麻烦,每次分析时内标物和试样都要准确称量,有时寻找合适的内标物也有困难。外标法简便,但进样量要求十分准确,要严格控制在与标准物相同的操作条件下进行,否则造成分析误差,得不到准确的测量结果。 内标与外标都是定量的一种方法而已,至于哪一种方法好与不好不能一概而论,做不同的分析,面对着不同的要求,再加上分析成本分析效率等等问题,我想简单而有效进行定量分析来满足要求才是最重要的。 1、以前做过很多医药、农药中间体的芳香族卤代化合物的常量定量分析,没有自
气相色谱定量分析中内标法和标准曲线法有什么区别?感觉差不多,都得绘制标准曲线,这两个曲线有什么区别?
国标5009.162有两个内标六氯苯和灭蚁灵 哪个化合物对应哪个内标并没有提 大家都怎么划分内标的呢
看文章,总是看到质谱中分子量的偏差(MASS ERROR)一般是ppm, 但一直不清楚是怎么计算的,谁懂这方面,能不能给讲解一下,谢谢了
请教各位大神! 我们在进行[url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]方法验证时,遇到问题:1.检测限和定量限有没有必要做? 2.线性如何做,有必要从定量限开始做吗? 2个问题参考以下方法 具体方法介绍如下:内标法 待测物质是A物质 内标物是B物质 产品A物质的规格限为3.5~5.4(备注:检测结果低于3.5为不合格,高于5.4也不合格) 线性我们做的是规格限的60%~150%(做线性时(只用A物质标液,没有用到内标物,单独考察A物质峰面积和浓度关系),是否合理。
[color=#333333]质谱分析中分子离子为什么一定是奇电子离子[/color]
内标法定量时没有样品标准物怎么办?如:检测到一样品中有10种成分,其中有5种能买到标准物采用内标法定量。另外5种买不到标准物该如何定量呢?结构相近的物质在气相色谱质谱联用中气化和电离化程度一样吗?可不可以用结构相近的能买得到的标准物定量?如,用丁酸的相对校正因子定量戊酸(假设戊酸买不到标准物)
选一与欲测组分相近但能完全分离的组分做内标物(当然是样品中没有的组分),然后配制欲测组分和内标物的混合标准溶液,进样得相对校正因子。再将内标物加入欲测组分的样品中,进样后测得欲测组分和内标物的定量参数。用内标法公式计算即可。 内标法是将一定量的纯物质作内标物,加入到准确称量的试样中,根据被测试样和内标物的质量比及其相应的色谱峰面积之比,来计算被测组分的含量。选择内标物有4个要求:1.内标物应是该试样中不存在的纯物质;2.它必须完全溶于试样中,并与试样中各组分的色谱峰能完全分离;3.加入内标物的量应接近于被测组分;4.色谱峰的位置应与被测组分的色谱峰的位置相近,或在几个被测组分色谱峰中间。内标法的优点是测定的结果较为准确,由于通过测量内标物及被测组分的峰面积的相对值来进行计算的,因而在一定程度上消除了操作条件等的变化所引起的误差。内标法的缺点是操作程序较为麻烦,每次分析时内标物和试样都要准确称量,有时寻找合适的内标物也有困难。外标法简便,但进样量要求十分准确,要严格控制在与标准物相同的操作条件下进行,否则造成分析误差,得不到准确的测量结果。 内标与外标都是定量的一种方法而已,至于哪一种方法好与不好不能一概而论,做不同的分析,面对着不同的要求,再加上分析成本分析效率等等问题,我想简单而有效进行定量分析来满足要求才是最重要的。 1、以前做过很多医药、农药中间体的芳香族卤代化合物的常量定量分析,没有自动进样器,用外标法定量,确实重现性与稳定性非常差,结果经常受到搞合成同事的质疑。其实,仔细分析原因不一定就是外标法不适合这种定量分析,首先我们的实验室仪器和手段是否调整到一种稳定而合理的状态了,比如,衬管是否洁净,玻璃棉的位置是否合适恰当(能否使样品尽可能的汽化)、汽化温度是否合适、色谱峰形是否对称(也就是样品与色谱柱健合相是否匹配)、附近有没有其它色谱峰的干扰、选用什么进样方式(如快速进样还是热针进样)等等因素的影响都需要考虑,如果这些因素都考虑了,按照GMP方法验证对于精密度的要求,同一样品进6针以上的RSD和配制6个样品的定量结果RSD都能满足小于1.5%的要求,那么这个方法用外标法就是完全适用的,但是前面的影响因素是一定要都考虑到的,否则谈论这个方法是否适用就有失偏颇了。在做过的许多出口产品的定量分析方法当中有许多是一些医药公司提供的比较完善而验证过的方法,内标与外标都有(他们用的都是自动进样)精密度都能满足RSD小于1.5%的要求,当一个方法能够满足测试要求的时候,无论内标外标,都是可行的,当然有一个分析成本和分析时间的问题,内标的成本和控制溶液、样品溶液的配制当然要比外标要高和麻烦一些了。而有些时候,可能受你实验室现有仪器和附属设备的影响,达不到一定的要求,而还必须进行定量分析,有时外标的结果可能就要差一些,这时,你可能就要考虑用内标法了,可以排除手动进样的误差、分流歧视的影响、包括一些未知因素平行误差的影响,这时内标可能就显示出它的优势来了。 2、上面已经提到当做方法验证的时候,当同一样品配制6个样品溶液用所选用的外标法进行定量的时候,RSD都满足1.5%的要求时,也分为两种情况,小于1%和大于1%小于1.5%。如果RSD的结果小于1%,那这个方法就没有什么可以怀疑的了;如果RSD的结果大于1%而在1.5%略低一些的范围活动时,这个方法的可行性就将受到质疑,毕竟这是方法验证,你就要考虑上面1所提到的影响因素的影响了,如果排除掉以上的影响因素,RSD还是在1.5%附近,就要尝试内标了,如果内标结果的RSD很好,就证明你的这个方法受实验条件的影响很大,只能用内标了,或者干脆将原方法做大的变动,再尝试用外标法测试。 3、而对于微量分析,比如农药和兽药残留的分析、环境分析等,根据不同的限量标准要求对于精密度的要求也比常量分析的要求要宽松的多,RSD有时可以允许达到10%甚至更高,这时可能外标法有更大的应用空间。 4、单从精密度方面去考虑,排除其它成本和效率的因素,个人认为还是内标优于外标。曾经做过一个中间体二氨基丙醇的常量定量分析,以二乙醇胺为内标,RTX-5 amine(碱改性) 15m*0.32mm*1.0um色谱柱分析,将配制好的控制溶液(含有内标物)自动进样器进6针,目的物(二氨基丙醇)与内标物(二乙醇胺)峰面积比率的RSD为0.18%,而只对这六针样品的目的物峰(二氨基丙醇)面积求RSD,结果为0.71%,通过这一实例的结果大家就会发现到底哪个方法精密度更好了,当然是内标更好了。当然这个化合物的检测方法最后根据上面的验证数据用内标和外标定量都是可以的,实验室可以自由选择。但内标与外标精密度结果的差异是显然存在的事实。 结论:应用外标法能够满足要求,首选还是外标法了,毕竟简单而省事。对于精密度要求比较高、结果准确度会产生重大影响、实验室条件不是很理想的等等条件下,用内标法还是必要的。无论应用那种方法,方法的验证和确认都是很重要的,只要是按照程序经过验证和确认的方法,都有其应用的空间的
目前想用Thermo Fisher的四级杆-静电轨道离子阱质谱做一个药物单,二,三磷酸核苷酸分析。文献报道是用同位素做内标,是他们实验室自己合成的,买不到现成的。用阴离子,MRM模式来做。想问,如果没有内标的话,只有该药物的单,二,三磷酸核苷酸的标准品的话,能够定量吗?第一次发帖求助,先谢谢大家的热心帮助!







 我要推广仪器
我要推广仪器
 下载APP
下载APP