在满足灵敏度要求的前提下,减少进样体积有助于消除溶剂效应。如果进样体积没有降低空间,又不想调整分析方法,可以使用消除溶剂效应小工具。它的工作原理是在色谱柱与进样器之间增加一个工具,把样品与流动相提前预混充分,再进入色谱柱。可以有效改善溶剂效应。
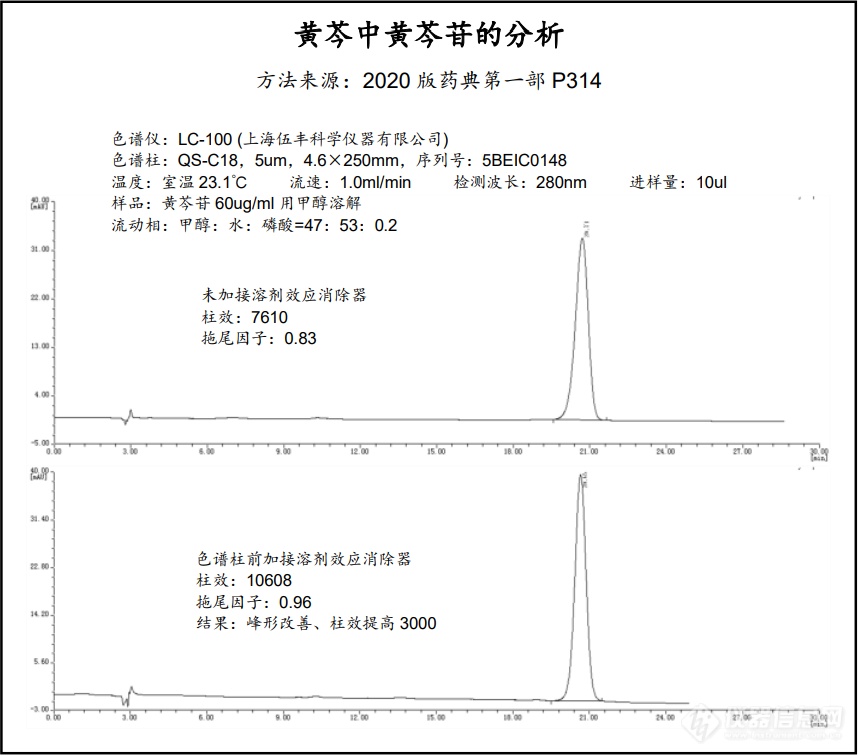
[font=&][/font][font=&] 自峰形前延抑制器发布以来,在许多用户的应用实践中发现了它的一些新能力,超出了最初的设计范围,它不仅有抑制峰形前延的作用,还有稳定保留时间,防止基线漂移,对传统手段无法解决的前延状况(样品已用流动相溶解,传统方案无法解决)仍然表现出改善峰形、提高柱效和分离度的奇效。针对新的情况,本公司对溶剂效应的产生原理进行了更深入的研究,并结合本产品的结构特点对其消除溶剂效应的机理进行了更新层次的探讨和解释,原有的名称“峰形前延抑制器”已不足以概括它的功能,因此更名为“溶剂效应消除器”,并对其硬件进行了升级,原有的“峰形前延抑制器YZQ-001”不再生产,取而代之的是新一代的溶剂效应消除器“Solvnt-Smoother Solvs-AB”,货号:Solvs-AB。[/font][font=&][/font][font=&]以下是用伍丰 LC-100进行分析的应用图谱,2020版药典方法,黄芩中黄芩苷的分析。[/font][img=,690,607]https://ng1.17img.cn/bbsfiles/images/2021/06/202106071030331319_8051_2568233_3.png!w690x607.jpg[/img]
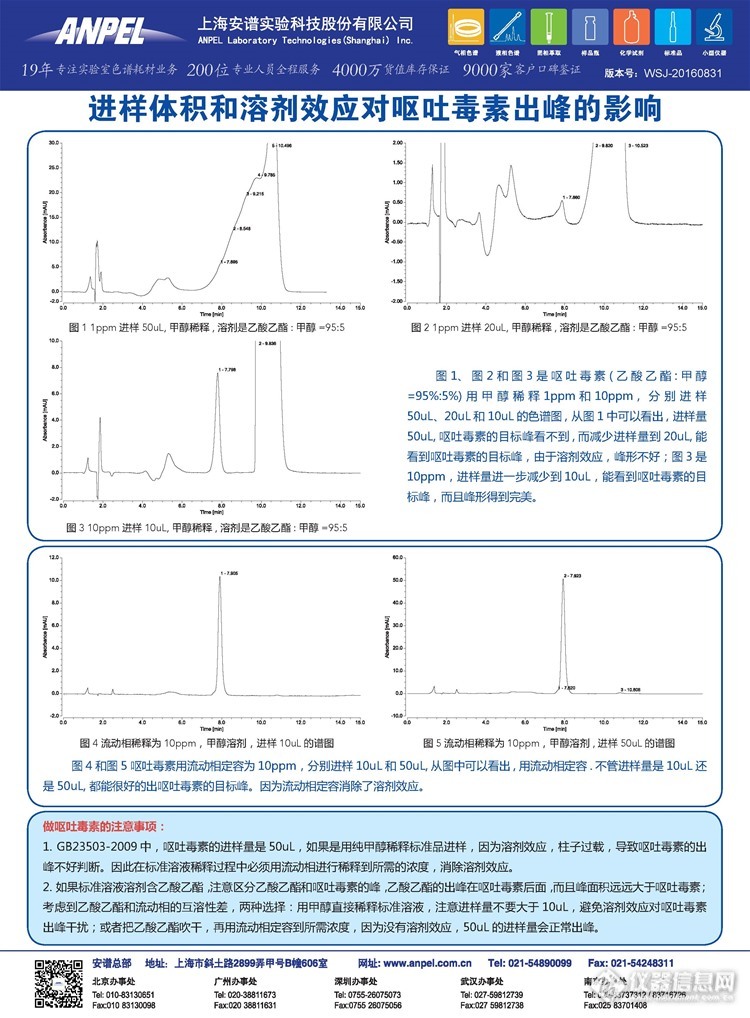
进样体积和溶剂效应对色谱出峰的影响巨大,有时候,因为溶剂效应,目标物的出峰会莫名其妙,大家工作中有默有?下面这个例子比较典型,食品中呕吐毒素的检测:因为进样量为50UL,溶剂效应明显,用流动相去溶解你的样品,可以消除溶剂效应:http://ng1.17img.cn/bbsfiles/images/2016/09/201609021451_608078_1835694_3.jpg
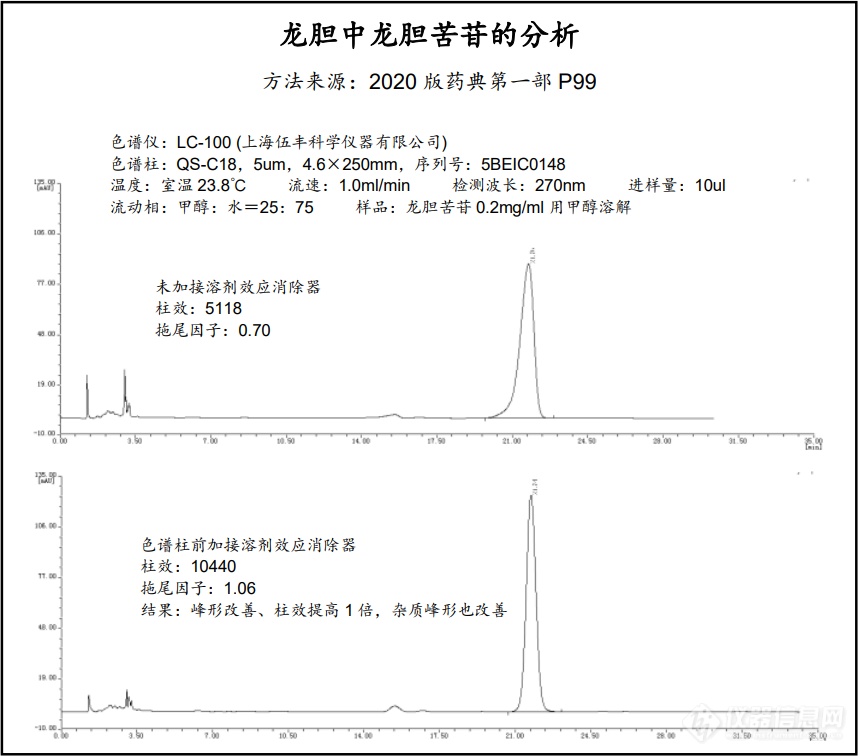
自峰形前延抑制器发布以来,在许多用户的应用实践中发现了它的一些新能力,超出了最初的设计范围,它不仅有抑制峰形前延的作用,还有稳定保留时间,防止基线漂移,对传统手段无法解决的前延状况(样品已用流动相溶解,传统方案无法解决)仍然表现出改善峰形、提高柱效和分离度的奇效。针对新的情况,本公司对溶剂效应的产生原理进行了更深入的研究,并结合本产品的结构特点对其消除溶剂效应的机理进行了更新层次的探讨和解释,原有的名称“峰形前延抑制器”已不足以概括它的功能,因此更名为“溶剂效应消除器”,并对其硬件进行了升级,原有的“峰形前延抑制器YZQ-001”不再生产,取而代之的是新一代的溶剂效应消除器“Solvnt-Smoother Solvs-AB”,货号:Solvs-AB。以下是用伍丰 LC-100进行分析的应用图谱,2020版药典方法,中药龙胆中龙胆苦苷的分析。[img=,690,607]https://ng1.17img.cn/bbsfiles/images/2021/06/202106070833126994_5688_2568233_3.png!w690x607.jpg[/img]
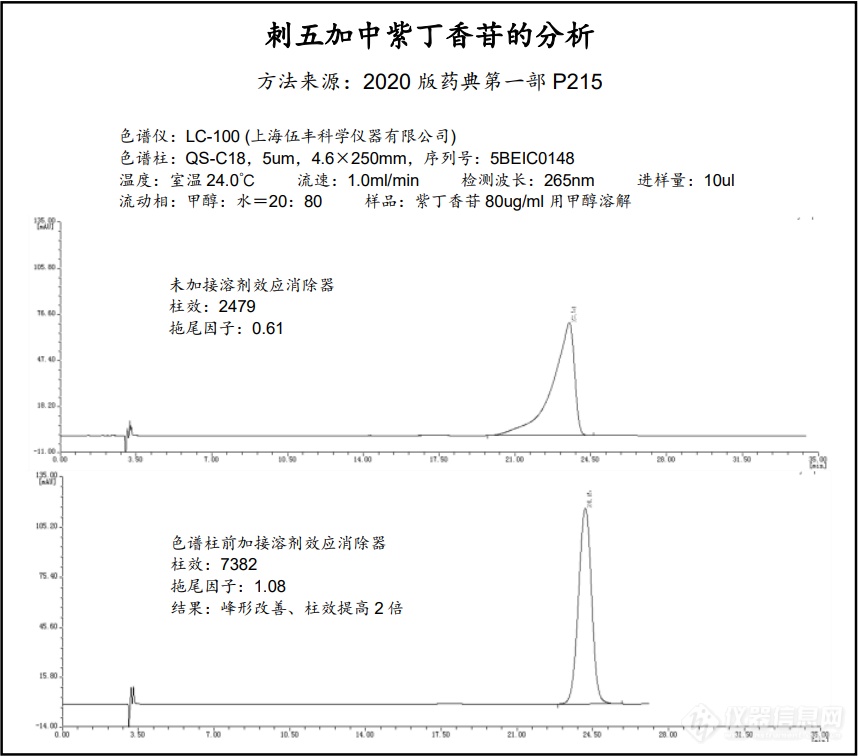
自峰形前延抑制器发布以来,在许多用户的应用实践中发现了它的一些新能力,超出了最初的设计范围,它不仅有抑制峰形前延的作用,还有稳定保留时间,防止基线漂移,对传统手段无法解决的前延状况(样品已用流动相溶解,传统方案无法解决)仍然表现出改善峰形、提高柱效和分离度的奇效。针对新的情况,本公司对溶剂效应的产生原理进行了更深入的研究,并结合本产品的结构特点对其消除溶剂效应的机理进行了更新层次的探讨和解释,原有的名称“峰形前延抑制器”已不足以概括它的功能,因此更名为“溶剂效应消除器”,并对其硬件进行了升级,原有的“峰形前延抑制器YZQ-001”不再生产,取而代之的是新一代的溶剂效应消除器“Solvnt-Smoother Solvs-AB”,货号:Solvs-AB。 以下是用伍丰 LC-100进行分析的应用图谱,2020版药典方法,刺五加中紫丁香苷的分析。[img=,690,607]https://ng1.17img.cn/bbsfiles/images/2021/06/202106070839279362_6719_2568233_3.png!w690x607.jpg[/img]
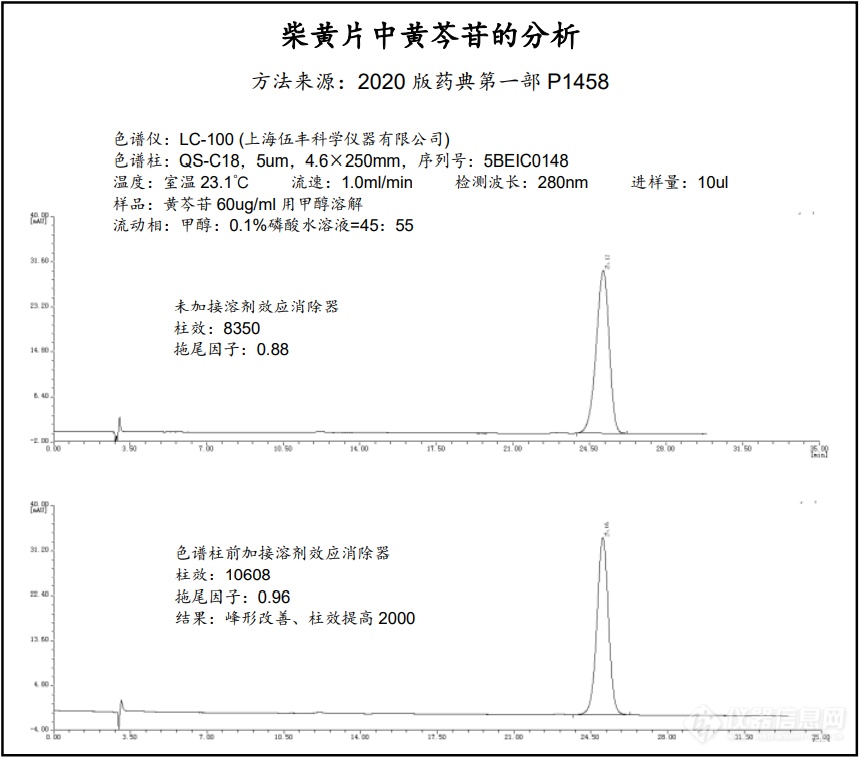
[font=&] 自峰形前延抑制器发布以来,在许多用户的应用实践中发现了它的一些新能力,超出了最初的设计范围,它不仅有抑制峰形前延的作用,还有稳定保留时间,防止基线漂移,对传统手段无法解决的前延状况(样品已用流动相溶解,传统方案无法解决)仍然表现出改善峰形、提高柱效和分离度的奇效。针对新的情况,本公司对溶剂效应的产生原理进行了更深入的研究,并结合本产品的结构特点对其消除溶剂效应的机理进行了更新层次的探讨和解释,原有的名称“峰形前延抑制器”已不足以概括它的功能,因此更名为“溶剂效应消除器”,并对其硬件进行了升级,原有的“峰形前延抑制器YZQ-001”不再生产,取而代之的是新一代的溶剂效应消除器“Solvnt-Smoother Solvs-AB”,货号:Solvs-AB。[/font][font=&]以下是用伍丰 LC-100进行分析的应用图谱,2020版药典方法,柴黄片中黄芩苷的分析[/font][font=&][img=,690,607]https://ng1.17img.cn/bbsfiles/images/2021/06/202106070957471571_4563_2568233_3.png!w690x607.jpg[/img][/font]
溶剂效应是样品溶液的溶剂强度强于流动相溶剂强度时可能会造成的峰展宽、峰分叉现象。现象色谱图上较早洗脱的峰扭曲变形或者开叉,与此同时较晚洗脱的峰则较为尖锐与对称,这些现象显示一个比较特殊的起因――样品溶液的溶剂很可能强于流动相。例如样品溶液的溶剂是100%乙腈(100%的强溶剂),而流动相的组成则较弱,18%的乙腈与72%的水。第一个峰是开叉的,并且与第二个峰相比,明显地变宽了。当样品溶液的溶剂变成流动相时,所有的峰形都改善了,且变得尖锐。解释当样品进样时,有可能出现峰展宽,最佳的样品溶液组成和体积将会保持在10%甚至更低,在这个例子里,当样品溶剂与流动相溶剂强度不同时,换句话来说,也就是样品未用流动相溶解,因此,有些样品分子溶解在强溶剂(100%ACE),并随强溶剂流过柱子,而有些则溶解在流动相中,从而导致峰分叉。当样品与流动相强度相差较小,进样影响也会小,第一个峰可能会宽于第二个峰,而当这种展宽导致必要的分离度降低时,这样情况应引起注意,例如使用一根短柱,和5UL进样,这与最佳进样体积4UL相近,用了极性更强的溶剂导致分离度明显的降低。避免的方法尽量用流动相来溶解样品,对于梯度洗脱,采用初始的流动相比例。对于在流动相中溶解度小,必须用强溶剂的时候,减少进样体积以消除溶剂效应的影响。版面相关帖子汇总:1、正确分析问题才能有效解决问题——不可忽视的溶剂效应2、出现溶剂效应了,请高人推荐流动相方案3、溶剂效应会影响峰面积吗?4、氨基酸衍生后样品溶液溶剂效应问题5、【原创】容易忽略的溶剂效应6、关于溶剂效应7、进样的溶剂效应8、用流动相定容样品和稀释储备液是否可以完全避免溶剂效应?9、检测红景天甙遇到了问题,分享下解决经历
溶剂效应是指溶剂对于反应速率,平衡甚至反应机理的影响。样品溶液的溶剂强度强于流动相溶剂强度时可能会造成的峰展宽、峰分叉现象。现象:色谱图上较早洗脱的峰扭曲变形或者开叉,与此同时较晚洗脱的峰则较为尖锐与对称,这些现象显示一个比较特殊的起因――样品溶液的溶剂很可能强于流动相。例如样品溶液的溶剂是100%乙腈(100%的强溶剂),而流动相的组成则较弱,18%的乙腈与72%的水。第一个峰是开叉的,并且与第二个峰相比,明显地变宽了。当样品溶液的溶剂变成流动相时,所有的峰形都改善了,且变得尖锐。解释:当样品进样时,有可能出现峰展宽,最佳的样品溶液组成和体积将会保持在10%甚至更低,在这个例子里,当样品溶剂与流动相溶剂强度不同时,换句话来说,也就是样品未用流动相溶解,因此,有些样品分子溶解在强溶剂(100%ACE),并随强溶剂流过柱子,而有些则溶解在流动相中,从而导致峰分叉。当样品与流动相强度相差较小,进样影响也会小,第一个峰可能会宽于第二个峰,而当这种展宽导致必要的分离度降低时,这样情况应引起注意,例如使用一根短柱,和5UL进样,这与最佳进样体积4UL相近,用了极性更强的溶剂导致分离度明显的降低。避免的方法:尽量用流动相来溶解样品,对于梯度洗脱,采用初始的流动相比例。对于在流动相中溶解度小,必须用强溶剂的时候,减少进样体积以消除溶剂效应的影响。
在开启分流阀的那一瞬间,气化室大部分溶剂气体(包括5%的样品组分)从分流口流出。为什么大量溶剂气体却只有5%样品,在气化室均匀气化的,如果进入色谱柱95%的样品,那溶剂也应该进去了95%啊,还怎么消除溶剂拖尾
比如样品的定容的溶剂不一样,峰形也会不一样,这就需要我们选择合适的溶剂来溶解。不知道气相色谱是否存在存在溶剂效应?
各位老师,我现在在重复一个Usp的液相分析方法,流动相是2%甲醇_三氟乙酸水溶液,稀释液是60%的甲醇,水溶液里面有碱,运行后第一个色谱峰严重分叉,这个峰出在四分钟,但是还有一个峰出在6分钟确峰形良好,调整稀释液为20%甲醇,分叉改善,峰形良好,表明一个稀释液和色谱流动相有严重问题,但是稀释液改了后样品的溶解性和稳定性都有问题,所以稀释液不能改,而且按目前的注册风向,改Usp的方法是比较忌讳的。但是作为Usp方法,应该是验证复核过的,为什么会出现这样的问题呢?还是我们的色谱系统不行?我听说有一款溶剂效应消除器,哪里有卖?
作为气相色谱的新手,对溶剂的效应十分困惑,谁能详细给我讲述一下溶剂效应呢?先说说我自己的理解,溶剂气话后进入色谱柱,冷凝 ,如果溶剂的极性和固定相相差大,将不能和固定相很好的结合,这将导致溶剂分布较宽,由于样品分布在溶剂里面样品也会展宽。其次,冷凝是部分冷凝怎么办,会不会一部分液态保留了,气态的跑了。希望高手能为我解惑。
样品溶液的溶剂强度强于流动相溶剂强度时可能会造成的峰展宽、峰分叉现象。。 现象 色谱图上较早洗脱的峰扭曲变形或者开叉,与此同时较晚洗脱的峰则较为尖锐与对称,这些现象显示一个比较特殊的起因――样品溶液的溶剂很可能强于流动相。例如样品溶液的溶剂是100%乙腈(100%的强溶剂),而流动相的组成则较弱,28%的乙腈与72%的水。第一个峰是开叉的,并且与第二个峰相比,明显地变宽了。当样品溶液的溶剂变成流动相时,所有的峰形都改善了,且变得尖锐。 解释 当样品进样时,有可能出现峰展宽,最佳的样品溶液组成和体积将会保持在10%甚至更低,在这个例子里,当样品溶剂与流动相溶剂强度不同时,换句话来说,也就是样品未用流动相溶解,因此,有些样品分子溶解在强溶剂(100%ACE),并随强溶剂流过柱子,而有些则溶解在流动相中,从而导致峰分叉。 当样品与流动相强度相差较小,进样影响也会小,第一个峰可能会宽于第二个峰,而当这种展宽导致必要的分离度降低时,这样情况应引起注意,例如使用一根短柱,和5UL进样,这与最佳进样体积4UL相近,用了极性更强的溶剂导致分离度明显的降低。 避免的方法 尽量用流动相来溶解样品,对于梯度洗脱,采用初始的流动相比例。对于在流动相中溶解度小,必须用强溶剂的时候,减少进样体积以消除溶剂效应的影响。 依稀记得似乎发过一次了,如果是,版主可以删帖哦
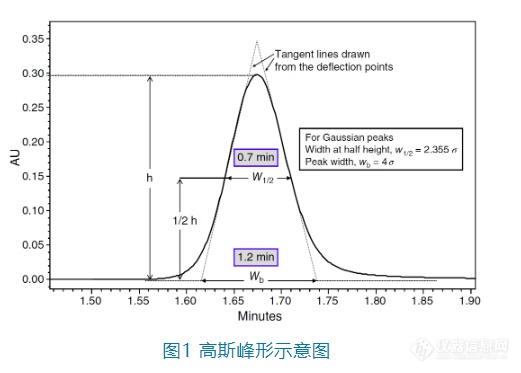
[size=14px]峰形问题是反相色谱分析中最常遇到的问题之一。造成峰形问题的原因有很多,[/size][size=14px]本次我们重点讨论在反相色谱中由于稀释剂与初始流动相的差异造成的峰形问题,也就是常说的“溶剂效应”及其内在原因和应对方法。[/size][size=16px][b][color=#007aaa]1. 为什么我们需要好的色谱峰形?[/color][/b][/size][size=14px]色谱分析是一种非常追求准确度的分析手段,理想的峰形是高斯(Gaussian)峰形,也就是呈正态分布,左右对称。但是在实际情况下,色谱峰或多或少都会有点前延(fronting)或者拖尾(tailing),甚至完全不成单峰等,这样[/size][size=14px][color=#7b0c00]不仅影响分析人员看到图谱时的心情,也会降低色谱峰的灵敏度、峰面积的积分准确度以及色谱峰之间的分离度[/color][/size][size=14px]。[/size][align=center][img=,521,373]https://ng1.17img.cn/bbsfiles/images/2022/12/202212011443520791_9645_3237657_3.png!w521x373.jpg[/img][/align][align=center][/align][size=16px][b][color=#007aaa]2. 什么是理想的样品稀释剂?[/color][/b][/size][size=14px]在分析HPLC中,往往建议稀释剂为起始流动相(但在制备HPLC中,使用比流动相更弱的稀释剂更利于更大的进样体积),以避免对分离分析产生不利影响。但是在实际的工作中,样品的预处理往往使得其稀释剂和起始流动相有了很大的差异,也有一些分析方案中会建议通过蒸发除去样品预处理中的溶剂后再用起始流动相溶解样品。但是这种额外的步骤很费时间,甚至超过了HPLC分析的时间,只有在万不得已的时候才选择。也有基于其他考虑(如溶解性、样品稳定性等)而选择和流动相不一致的溶剂作为稀释剂。但是[/size][size=14px][color=#7b0c00]不管是何种稀释剂,只要能稳定溶解足够的样品,且对峰形等不会产生不利影响,就是理想的稀释剂[/color][/size][size=14px]。[/size][size=16px][b][color=#007aaa]3. 溶剂效应的原因有哪些?如何应对?[/color][/b][/size][size=15px][b][color=#007aaa]3.1 稀释剂与流动相的洗脱强度不匹配[/color][/b][/size][size=15px][b][color=#007aaa][/color][/b][/size][size=14px]当稀释剂的洗脱强度强于起始流动相时,也就是有机相(%B)的比例高于流动相时,我们可以估算出保留的改变(Δ[i]k[/i]):[/size][align=center][img=,203,75]https://ng1.17img.cn/bbsfiles/images/2022/12/202212011445102816_8758_3237657_3.png!w203x75.jpg[/img][/align][size=14px]其中[i]S[/i]和每种溶质的特性有关,[i]S[/i] ≈ 0.25 MW[sup]0.5[/sup],MW为分子量。如果我们选择了一种典型的400 Da的小分子,当B%变化10%([/size][size=14px]即[i]Φ[/i]=0.1[/size][size=14px])时[i],S[/i] ≈ 0.25×400[sup]0.5[/sup]=5,Δ[i]k[/i] ≈ 10[sup]5×0.1[/sup] ≈ 3.2。这意味着B相的10%的增加或者降低会导致三倍的的保留因子K的降低或者增加,也就是常说的“三倍规则”。如果一个样品的稀释剂比流动相强,进样的溶液在跑过柱子的过程中,它会和流动相以相同的流速移动,但是其中的样品分子的移动速度会比溶解在流动相时更快,进样成分会被流动相逐渐稀释直到和流动相完全一致。我们把进样的液体形象地称为“样品塞”,紧跟“样品塞”的流动相会稀释它,因此尾部的溶质分子的前进速度会慢下来,以正常的洗脱速度前进。[/size][size=14px][color=#7b0c00]虽然这些过程发生的速度很快,但是如果稀释剂足够强、进样体积足够大,还是会有明显的峰展宽[/color][/size][size=14px]。[/size][size=14px]Dolan作了图来更加直观地说明这种现象,在图2中每个水平放置的长条代表充满流动相(用小圆黑点表示)的色谱柱。在上面三个例子中,进到流动相的样品用棋盘格表示,随着时间的流逝从状态A逐步变到状态B和C,样品流经色谱柱,发生了稍微的峰展宽。在下面三个例子中,样品溶解在更强的溶剂中(斜线表示),A'为刚进样后的状态,“样品塞”占据的体积和A一样,因为进样量相同;在B'中,由于“样品塞”里的溶剂比流动相更强,溶解其中的样品分子会以比正常情况更快的速度通过色谱柱,与此同时尾部被流动相稀释的部分溶质分子的通过速度降低至正常速度(棋盘格部分),这样即使“样品塞”更窄(也就是进样量更少),由于前后端的稀释,样品扩散的空间也会更大;在C'中,“样品塞”基本都被稀释了,但是峰展宽一直在发生,直到所有样品都溶解在流动相中。[/size][align=center][/align][align=center][img=,690,328]https://ng1.17img.cn/bbsfiles/images/2022/12/202212011445266041_6974_3237657_3.png!w690x328.jpg[/img][/align][align=center][/align][size=14px]在某些更极端的条件下,稀释剂的强度太大还可能导致色谱峰出现裂分甚至完全不成峰形。[/size][align=center][/align][size=14px]当由于稀释剂由于和流动相的溶剂强度不匹配而造成峰形异常时,该如何应对?[/size][size=14px][color=#7b0c00]1. 降低稀释剂的洗脱强度;[/color][/size][size=14px][color=#7b0c00]2. 降低样品的进样体积。进样体积越小,样品就越容易被流动相稀释掉。Dolan实验室尝试过在反相流动相中进样1 μL以甲苯为溶剂的样品时,可以得到满意的结果。[/color][/size][size=14px][color=#7b0c00]但是稀释剂与流动相的洗脱强度不匹配并不能解释所有情况[/color]。比如Tseing和Roger发现分析二羟基苯的异构体时,不管是将甲醇作为稀释剂,水作为流动相还是水作为稀释剂,甲醇作为流动相,最终都得到双峰,但是当稀释剂和流动相一致时,就可以得到单峰;Zapata和Garrido也发现在分析叶绿素时,当使用丙酮-水(69:31)为稀释剂,甲醇-水(95:5)为流动相时,虽然两者洗脱强度一致,但是叶素绿的峰为多峰。经过研究后发现这和稀释剂与流动相之间的[color=#7b0c00]黏度不匹配[/color]有关。[/size][size=15px][b][color=#007aaa]3.2 稀释剂与流动相的黏度不匹配[/color][/b][/size][size=14px]当稀释剂与流动相之间的黏度不一致时,会发生黏性指进(viscous fingering)这种流体力学不稳定现象。也就是低黏度的溶剂会如伸出手指一般向高粘度渗透,[/size][size=14px][color=#7b0c00]同时这种渗透是随机的,不可预测[/color][/size][size=14px]。如图3,黑色部分就是低黏度的溶剂,在更高粘度的背景溶剂中如树枝般随机“生长”。[/size][align=center][/align][align=center][img=,420,395]https://ng1.17img.cn/bbsfiles/images/2022/12/202212011445442312_5735_3237657_3.png!w420x395.jpg[/img][/align][align=center][/align][size=14px]在色谱分析中,稀释剂与流动相的黏度不匹配对谱图的影响不仅与稀释剂与流动相黏度的不匹配程度有关,也与样品溶质及样品溶剂的迁移速度有关。当黏度差异相同时,样品的溶质与溶剂在越短的时间内洗脱,且洗脱时间越接近时,对峰形的不利影响就越明显,这种现象在体积排阻色谱(SEC)中比较常见。[/size][size=14px]正如上面提到的这种“黏性指进”是随机的,因此[/size][size=14px][color=#7b0c00]往往连续进样时峰形不能重现(如图4),这也是判断到底是黏性不匹配还是上面所说的洗脱强度不匹配的重要依据[/color][/size][size=14px]。[/size][align=center][/align][align=center][img=,690,397]https://ng1.17img.cn/bbsfiles/images/2022/12/202212011446015690_3262_3237657_3.png!w690x397.jpg[/img][/align][size=14px]当由于稀释剂由于和流动相的黏度不匹配而造成峰形异常时,该如何应对?[/size][size=14px][/size][size=14px][color=#7b0c00]1. 尝试用初始流动相稀释样品,但这往往是正确的废话,因为很多情况是不能或不便用初始流动相稀释样品;[/color][/size][size=14px][color=#7b0c00]2. 降低进样量;[/color][/size][size=14px][color=#7b0c00]3. 选择更大的定量环,也就是增强在定量环中的预混,如图5。同样进样10 μL或者20 μL,当增加定量环的大小(从10 μL到20 μL或者从20 μL到50 μL)时,峰形可以明显改善。 [/color][/size][align=center][/align][align=center][img=,690,429]https://ng1.17img.cn/bbsfiles/images/2022/12/202212011446205122_5886_3237657_3.png!w690x429.jpg[/img][/align][size=14px][color=#7b0c00]4. 升高温度,可以在某些区间内拉平稀释剂和流动相之间的黏度差异,从而减少其不良影响,这在体积排阻色谱(SEC)中很常见;[/color][/size][size=14px][color=#7b0c00]5. 在半制备或者制备色谱中,常常增大进样量以提高效率,有时候由于溶解性问题也让使用起始流动相作为稀释剂不太现实,为了降低稀释剂和流动相由于黏度不匹配而造成分离的不良影响,强烈建议调节两者的黏度,一种可能的解决方案是在低黏度的一方中加入极少量的高黏度溶剂,比如DMSO或者环己醇。[/color][/size][size=15px][b][color=#007aaa]3.3 稀释剂与流动相的pH不匹配[/color][/b][/size][size=14px]我们在分析可离子化的化合物时,常常会基于分析物的[font=&]p[/font][i]K[/i][font=&]a[/font]选择不同pH的流动相,以实现最佳的分离效果。在很多情况下,样品稀释剂和流动相的pH并不一致,甚至相差好几个单位,比如样品在起始流动相条件下不稳定时,首先就应该考虑样品的稳定性。[/size][size=14px][color=#7b0c00]当稀释剂与流动相的pH不一致时,稀释剂在流经色谱柱时,会改变周围环境的pH,从而可能改变分析物的电离状态进而影响其保留行为[/color][/size][size=14px]。[/size][size=14px]为了更加直观地了解当样品稀释剂对流动相pH的影响,Stoll在HPLC系统中设计了一个pH的在线监测系统,可以实时测定色谱柱入口和色谱柱出口的pH值。实验中选择的苯甲酸为分析物,样品稀释剂为乙腈:1 mM或100 mM磷酸盐(pH 7)=13:87,流动相为乙腈:100 mM磷酸盐(pH 3.2)=23:77,色谱柱为50×4.6 mm的C18柱,流速为2 mL/min(其他详细色谱条件见参考文献[6])。图(a)为稀释剂中缓冲剂为1 mM pH 7的缓冲盐时,不同进样量时色谱柱入口处的pH随时间的变化图;图(b)为稀释剂中缓冲剂为100 mM pH 7的缓冲盐时,不同进样量时色谱柱入口处的pH随时间的变化图;图(c)为稀释剂中缓冲剂为1 mM pH 7的缓冲盐时,不同进样量时色谱柱出口处的pH随时间的变化图;图(d)为稀释剂中缓冲剂为100 mM pH 7的缓冲盐时,不同进样量时色谱柱入口处的pH随时间的变化图。其中蓝、紫、绿、红、粉曲线分别是进样量为1 μL、5 μL、15 μL、30 μL、100 μL的pH曲线图。[/size][align=center][/align][align=center][img=,690,423]https://ng1.17img.cn/bbsfiles/images/2022/12/202212011446451986_2628_3237657_3.png!w690x423.jpg[/img][/align][font=&][size=14px]在色谱柱入口端:[/size][/font][font=&][size=14px]从图(a)可以看出,即使稀释剂中的缓冲剂为1 mM的磷酸盐,当进样量为30或100 μL时也会在流动相中产生pH5的区域,而[/size][/font][font=&][size=14px][color=#7b0c00]当稀释剂中的缓冲剂为100 mM的磷酸盐时,见图(b),即使只进样5 μL,也会让流动相中产生pH值到6的区域[/color][/size][/font][font=&][size=14px]。[/size][/font][font=&][size=14px] [/size][/font][font=&][size=14px]在色谱柱出口端:[/size][/font][font=&][size=14px]我们可以看到其最大的pH值比色谱柱入口端监测的数据要低,这应该主要和样品的稀释剂的扩散和流动相对其的中和有关。当稀释剂中的缓冲剂浓度为1 mM时,如图C,样品稀释剂在色谱柱中充分扩散和被中和,监测到色谱柱后的pH基本都低于4,除非进样量达到100 μL时,才有少量的区域pH高于4;而[/size][/font][font=&][size=14px][color=#7b0c00]当样品稀释剂浓度为100 mM时,如图6D,当进样量为15 μL,30 μL和100 μL,会让色谱柱中很大的区域的pH都大于5[/color][/size][/font][font=&][size=14px]。[/size][/font][font=&][size=14px] [/size][/font][font=&][size=14px]而苯甲酸的p[i]K[/i]a为4.2,当色谱柱中的pH值由流动相的3.2升高时,让苯甲酸的电离程度也提高,从而让其保留时间降低,当pH值的变化比较剧烈时(如稀释剂的磷酸盐浓度为100 mM,进样量为15 μL或30 μL),色谱峰的保留时间显著减少,同时峰的拖尾变得更加严重,而当进样量为100 μL时,色谱峰会出现严重的变形和裂分现象,如图7。[/size][/font][font=&][size=14px][/size][/font][align=center][img=,690,289]https://ng1.17img.cn/bbsfiles/images/2022/12/202212011447093785_1060_3237657_3.png!w690x289.jpg[/img][/align][align=center][/align][font=&][size=14px]综合图6和图7可以看出,分析物的出峰行为和稀释剂对流动相pH的影响情况出现了非常强的相关性。我们可以看到当稀释剂与流动相的pH不匹配时,特别在稀释剂中缓冲液的缓冲能力比较强、进样量比较大的时,对于p[i]K[/i]a在特定范围内的分析物的出峰行为产生不可忽视的影响。相对应的,[/size][/font][font=&][size=14px][color=#7b0c00]降低稀释剂的缓冲能力、减少进样体积以及让稀释剂对流动相pH的影响范围尽量避开所关注的分析物的p[i]K[/i]a都是有效的方向[/color][/size][/font][font=&][size=14px]。[/size][/font][size=16px][b][color=#007aaa]全文总结[/color][/b][/size][size=14px]在液相色谱分析中,“溶剂效应”指由于样品的溶剂(稀释剂)与初始流动相的差异而对分析物的色谱行为造成的影响,但是其具体原因却不能一概而论。本文综合了现有的一些研究,总结了三种不同原因造成的溶剂效应,包括[/size][size=14px][color=#7b0c00]洗脱强度不匹配[/color][/size][size=14px]、[/size][size=14px][color=#7b0c00]黏度不匹配[/color][/size][size=14px]和[/size][size=14px][color=#7b0c00]pH不匹配[/color][/size][size=14px](当然,还有可能有其他的不匹配因素导致的溶剂效应,本文仅作抛砖引玉,如有错误或者遗漏,请在评论区指出)。当遇到具体的问题时,需要具体分析找出原因,并针对性地进行改善。[/size][size=14px][/size][size=14px]参考文献[/size][size=14px][1] Michael W. Dong HPLC and UHPLC for Practicing Scientists, 2nd Ed. [b]2019[/b], Wiley [/size][size=14px][2] J.W. Dolan, [i]LC-GC North Am. [/i]22 (2004) 26 [/size][size=14px][3] S. Keunchkarian et al.[i] J. Chromatogr. A[/i] 1119 ([b]2006[/b]) 20 [/size][size=14px][4] C.B. Castells et al. [i]J. Chromatogr. A [/i]805 ([b]1998[/b]) 55 [/size][size=14px][5] M. Czok et al. [i] J. Chromatogr. [/i]550 ([b]1991[/b]) 705 [/size][size=14px][6] D.R. Stoll et al.[i] LCGC Europe[/i] 33 ([b]2002[/b]) 74.[/size]
我在看一本气相色谱书,看到溶剂效应这个词,很是不懂,想请教各位老师

http://ng1.17img.cn/bbsfiles/images/2013/07/201307251656_453654_2764190_3.jpg各位前辈、这是书上看到的、、要是我使用的溶剂所产生的峰是在所有组分之后、会不会消除溶剂效应呢?谢谢

液相色谱中追求一个高柱效,高分离是我们做液相色谱追求的目标,但是往往存在很多问题,比如:柱效,分离,拖尾,峰展宽,峰变形,裂缝等等,有些容易解决,有些尝试很多途径都无法改善,当然可能会是方法本身的原因,但是也存在一些忽略的问题:比如本文中提到的溶剂效应,举二例说明: (1)盐酸小檗碱,方法见三黄片补充方法,左图为调整方法前液相色谱图,峰严重变形,我的做法是调大有机相比例,减小进样量,结果出来右边的图1.jpg (5.5 KB)http://ng1.17img.cn/bbsfiles/images/2010/12/201012181618_268050_2019107_3.jpg2010-6-23 23:353.jpg (5.31 KB)2010-6-23 23:36 (2)阿莫西林,方法见中国药典二部,左两图为失败方法,右图为调整溶剂的图:需要说明的是我们的是多元泵,也就是作为溶剂的流动相为量筒配置,试验过程中调整了上机的流动相(有机相减少),而作为溶剂的未调。我的做法是将溶剂的流动相调比例至比上机流动相低5个比例。结果见右图。 2.jpg (9.26 KB)http://ng1.17img.cn/bbsfiles/images/2010/12/201012181618_268051_2019107_3.jpg2010-6-23 23:35 原因,第一例溶剂为甲醇,流动相为乙腈与离子对混合液,第二例溶剂有机相比例比流动相高,共同点为溶剂强度比流动相强。 解析:液相色谱一般进样量为10微升,柱子中局部流动相量比较小(没具体算),当样品溶液进入柱子,10微升体积会改变局部的流动相环境(强度),从而可能出现两种保留行为,反应在图谱上就是变形,如拖尾,裂缝,肩峰。 希望对做液相色谱的朋友有用。
进纯物质和进用溶剂稀释过的样品出峰时间会有差异,特别是多组分好像更加明显,但是又不是所有的组分出峰时间都有改变。这是因为有溶剂效应么?
求助各位同仁: 原先从文献上看到一个术语———“反溶剂效应”。 配有一张色谱图。 混合物,主峰之前的一个色谱峰,出现了峰宽展宽。 不知道具体是个什么过程,有没有详细的理论讨论呢? 多谢大家,不吝赐教。
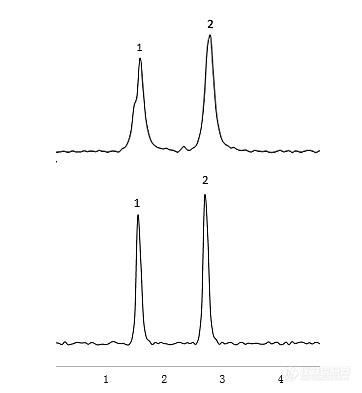
[b][font=宋体][back=white]问题描述:“溶剂效应”会导致哪些异常峰型?如何避免溶剂效应?在[/back][/font][back=white]HPLC[/back][font=宋体][back=white]分析中样品溶剂的选择与流动相有什么关系?[/back][/font][font=宋体][back=white]解答:[/back][/font][/b][font=宋体][back=white]([/back][/font][back=white]1[/back][font=宋体][back=white])当样品溶液的溶剂强度强于流动相溶剂强度时可能会导致峰前端展宽、峰分叉,即色谱图上较早洗脱的峰扭曲变形或者开叉,与此同时较晚洗脱的峰则较为尖锐与对称,这种现象一般称之为“溶剂效应”。[/back][/font][font=宋体][back=white]([/back][/font][back=white]2[/back][font=宋体][back=white])这里的强溶剂可以理解为洗脱能力强的溶剂,色谱常用的有机相是甲醇与乙腈,纯乙腈就是洗脱能力最强的溶剂。流动相的洗脱能力越强,在其他条件不变的情况下,出峰越快,保留时间越短。[/back][/font][font=宋体][back=white]([/back][/font][back=white]3[/back][font=宋体][back=white])液相色谱分析中产生“溶剂效应”的原因主要有:[/back][/font][back=white]a.[/back][font=宋体][back=white]样品溶液的溶剂强于流动相。如图[/back][/font][back=white]5-5[/back][font=宋体][back=white]所示,当样品溶液的溶剂是[/back][/font][back=white]100%[/back][font=宋体][back=white]乙腈(强溶剂),而流动相组成较弱([/back][/font][back=white]18%[/back][font=宋体][back=white]乙腈[/back][/font][back=white]/72%[/back][font=宋体][back=white]水):第一个峰是开叉的,并且与第二个峰相比,明显地变宽了(图[/back][/font][back=white]5-5[/back][font=宋体][back=white]上);当样品溶液的溶剂变成流动相时,所有的峰形都改善了,且变得尖锐(图[/back][/font][back=white]5-5[/back][font=宋体][back=white]下)。这是因为当样品进样时,有可能出现峰展宽,最佳的样品溶液组成和体积将会保持在[/back][/font][back=white]10%[/back][font=宋体][back=white]甚至更低,在这个例子里,当样品溶剂与流动相溶剂强度不同时,此时有些样品分子溶解在强溶剂中,并随强溶剂流过柱子,而有些则溶解在流动相中,从而导致峰分叉。[/back][/font][align=center][img=,362,393]https://ng1.17img.cn/bbsfiles/images/2021/03/202103221545160685_5875_3389662_3.jpg!w362x393.jpg[/img][/align][align=center][i][font=宋体]图[/font]5-5 [font=宋体]样品溶剂对组分峰型影响[/font][/i][/align][back=white]b.[/back][font=宋体][back=white]进样量比较大。[/back][/font][back=white]c.[/back][font=宋体][back=white]样品的[/back][/font][back=white]pH[/back][font=宋体][back=white]值与液相色谱流动相[/back][/font][back=white]pH[/back][font=宋体][back=white]悬殊,比如酸性流动相体系,样品[/back][/font][back=white]pH[/back][font=宋体][back=white]偏碱性([/back][/font][back=white]pH=10[/back][font=宋体][back=white]),这种情况下也很有可能发生“溶剂效应”。[/back][/font][font=宋体][back=white]([/back][/font][back=white]4[/back][font=宋体][back=white])其实上面三个原因归结起来就是一个原因:样品的溶剂与流动相存在一些不一样。当溶解样品的溶剂不同于流动相时,样品溶剂与流动相发生混合,样品溶剂被冲稀。如果进样溶剂之强度高于流动相,样品在瞬间会表现为在较强溶剂中,并以较快速度通过色谱柱,表现在色谱图上就是:色谱峰的保留时间缩短。当进样溶剂与流动相混合时,一部分分子会先与流动相混合,致使这些分子通过色谱柱的速度发生变化,使峰形扭曲,发生变形。[/back][/font][font=宋体][back=white]([/back][/font][back=white]5[/back][font=宋体][back=white])所以要解决这个问题的最佳方案就是,使用流动相的起始梯度溶解样品,如果由于溶解度的问题,不得不改用其余的溶剂,那就只能降低进样体积来解决。[/back][/font][font='微软雅黑','sans-serif'][color=black][back=white]领取更多《实战宝典》请进:[url]http://instrument-vip.mikecrm.com/2bbmrpI[/url][/back][/color][/font][font='微软雅黑','sans-serif'][color=black][back=white] [/back][/color][/font]

溶剂效应”会导致哪些峰形异常?如何避免溶剂效应?在HPLC分析中样品溶剂的选择和流动相是怎样的联系?为何建议使用流动相溶解样品?。。。。。一系列的问题都和样品溶剂的选择密切相关,如何解决真正的溶剂效应问题,又如何分辨哪些是非溶剂效应问题,不同的问题有不同的解决方法,有哪些前辈留下的实战经验可供分享呢?一起来看看吧!什么是溶剂效应?样品溶液的溶剂强度强于流动相溶剂强度时可能会造成的峰展宽、峰分叉现象。即色谱图上较早洗脱的峰前沿或者开叉,与此同时较晚洗脱的峰则较为正常的现象。例如样品溶液的溶剂是100%乙腈(100%的强溶剂),而流动相的组成则较弱,18%的乙腈与72%的水。第一个峰是开叉的,并且与第二个峰相比,明显地变宽了。当样品溶液的溶剂变成流动相时,所有的峰形都改善了,且变得尖锐。http://ng1.17img.cn/bbsfiles/images/2016/02/201602260950_585208_1624767_3.jpg可能发生溶剂效应的情况:出峰时间早;保留弱;进样量大。用流动相溶解样品能够得到很好的峰形 当溶解样品的溶剂不同于流动相时,样品溶剂与流动相发生混合,样品溶剂被冲稀。如果进样溶剂之强度高于流动相,样品在瞬间会表现为在较强溶剂中,并以较快速度通过色谱柱。表现在色谱图上就是∶色谱峰的保留时间缩短。`当进样溶剂与流动相混合时,一部分分子会先与流动相混合,致使这些分子通过色谱柱的速度发生变化,使峰形扭曲,发生变形。洗脱较早的色谱峰峰变形要比洗脱晚的色谱峰严重。解决进样溶剂问题的关键是使进样体积足够小,这样稀释过程会非常快;或使用比流动相弱的溶剂溶解样品。较弱的溶剂会使样品在色谱柱上发生浓缩,在某些情况下,色谱峰会比使用较强溶剂时窄一些。因此,在通常情况下,如果溶解样品的溶剂比流动相强,进样体积应不高于25mL。进样体积大小与进样溶剂与流动相之间的差别大小有关。这一差别很容易凭经验得到∶逐渐加大进样体积,直至发生峰变形现象。采用比发生峰变形时小一些的进样体积即可。问题中发生的峰变形就是因为溶解样品的溶剂甲醇强度远远高于流动相,因而得到了变形的、展宽的峰。如果使用水来溶解样品,溶解样品的溶剂弱于流动相,峰形会得以改善。在离子对色谱中,我们建议使用流动相来溶解样品,以最大程度地降低基线假峰的出现。样品溶剂分别为100%乙腈、流动相时峰形分析http://ng1.17img.cn/bbsfiles/images/2016/02/201602260951_585212_1624767_3.jpg上图中,色谱图上较早洗脱的峰扭曲变形或者开叉,与此同时较晚洗脱的峰则较为尖锐与对称,这些现象显示一个比较特殊的起因——样品溶液的溶剂很可能强于流动相。此种强溶剂效应的例子在左图中可见。此处的样品溶液的溶剂是100%乙腈(100%的强溶剂),而流动相的组成则较弱,18%的乙腈与72%的水。第一个峰是开叉的,并且与第二个峰相比,明显地变宽了。当样品溶液的溶剂变成流动相时,所有的峰形都改善了,且变得尖锐。见右图。为什么呢?这是因为当样品进样时,有可能出现峰展宽,最佳的样品溶液组成和体积将会保持在10%甚至更低,在这个例子里,当样品溶剂与流动相溶剂强度不同时,换句话来说,也就是样品未用流动相溶解,因此,有些样品分子溶解在强溶剂中,并随强溶剂流过柱子,而有些则溶解在流动相中,从而导致峰分叉.当样品与流动相强度相差较小,进样影响也会小,第一个峰可能会宽于第二个峰,而当这种展宽导致必要的分离度降低时,这样情况应引起注意,在左图中,使用一根短柱,和5μL进样,这与最佳进样体积4μL相近,用了极性更强的溶剂导致分离度明显的降低,从2.1降到1.5(如右图,分离度为2或更大是评估一个完善方法的一个必要参数,也是每天方法的验证参数,1.5只是一个基本的分离度,任何一个方法或一根柱子都必需满足这个条件,当进样为一倍时,也就是10μL时,分离度更一步降低,此方法就不行了。样品溶剂最好具有哪些特点比较好?http://ng1.17img.cn/bbsfiles/images/2016/02/201602260952_585213_1624767_3.jpg实战问答Q&A,绝对接地气!1.HPLC里面的溶剂效应是如何产生的,该如何避免?如何判断分叉的峰是否由溶剂效应造成,第一就是通过HPLC仪器比对一下两个劈叉峰的紫外吸收波谱,看看他们是否能完全一样的μV特征吸收,第二将进样体积调节到0.1μl看看,峰是否好转,如果满足上面两个条件几乎就可以肯定是100%的溶剂效应。产生原因1,样品溶液的溶剂很可能强于流动相。例如样品溶液的溶剂是100%乙腈(100%的强溶剂),而流动相的组成则较弱,18%的乙腈与72%的水产生原因2,就是进样量比较大,产生原因3,样品的pH值与HPLC流动相pH悬殊,比如酸性流动相体系,样品pH偏碱性(pH=10),这种情况下也很有可能发生其实上面三个原因归结起来就是一个原因,那就是样品的溶剂与流动相存在一些不一样,当样品进入流动相的时候,由于这种巨大的差异,一部分样品溶解进入了流动相,一部分还留在进样的溶剂里面,所以在HPLC上出现了保留的差异,所以要解决这个问题的最佳方案就是,使用流动相的起始梯度溶解样品,如果由于溶解度的问题,不得不改用其余的溶剂,那就只能降低进样体积来解决,比如 5μl,1μL。2.高效液相做专属性实验时,溶剂峰和目标峰出峰时间一样怎么办?分离的是氨基酸类物质,不是离子型化合物,流动相用的甲醇和水,也是用流动相配的溶剂,但是比例有少许差别,单进溶剂时确实有溶剂峰,用流动相配的样品可以和溶剂峰分开,我在做肠灌流实验,但是当样品进入动物体内灌流后,进样后就分不开了,我想把样品的出峰时间延长一些,认为能分开,但是不知道怎么调整,实在没办法。回答:问题1:你用的稀释剂是纯的有机相吗?稀释剂中有机相的比例一般尽可能与流动相一致,以减小溶剂效应.如果改变稀释剂仍不能改善,则说明你的色谱条件不合适,检测组分在色谱柱中几乎无保留.如果你的化合物是离子型的,一般需要使用缓冲盐,缓冲液pH值应在化合物pKa值正负2之外,以增强组分在色谱系统中的保留.如果不是离子型化合物,则减少有机相比例,以增加组分保留时间,避开溶剂峰.问题2:氨基酸类物质是两性化合物,出峰时间跟pH值有很大关系,样品经过动物体内后,pH值肯定是发生了变化的,样品的预处理就很重要了(这个你要请教其他高人,我没做过生物方面的药物分析)。如果你只是想单独延迟样品出峰时间,可以试着减少有机相比例,看能不能达到效果,如果不行,可能还是要用缓冲体系。3.醋酸曲安耐德用高效液相色谱测定,流动相为甲醇-水(60:40),ODS C18柱,紫外检测波长240nm,柱温30度。以前样品用甲醇溶解后直接进样,色谱峰各项参数正常。现在用甲醇溶解后进样,色谱峰变宽,必须用流动相溶解才能正常。仪器与色谱柱未更换,溶解样品的甲醇换成默克色谱纯甲醇也还是这样!什么原因?另外我需要测定的另一个中药中黄芩苷也出现了这种现象?我现在该怎么办?解答:溶剂效应是肯定的。溶剂效应的原理其实很简单:就是溶质的极性和流动相的极性不匹配,导致溶质被流动相包裹或部分包裹,不能充分分散并充分与C18交换,所以才导致峰展宽。你的醋酸曲安耐德极性并不是很强。我帮你查了它的log P,没查到,但根据我的经验,并根据其结构式我大概推算了一下,应该在2左右,应该属于中等偏弱极性的化合物(C26)。所以其在60:40这样含水流动相匹配度肯定不好。以前你做的没有问题,一个原因是上面各位所说的,溶解样品的甲醇可能含水量稍微高一些,比如大约含水2%(只是猜测哈),可能就不会出现溶剂效应,而这次用的甲醇含水量很低
近年来,质谱被广泛的应用于新药研发的各个领域,串联质谱以其特异性著称,因此,很多人认为既然串联质谱特异性强,就可以简化样品前处理或者可以缩短柱分离的时间。但最近越来越多的数据表明用质谱检测需要注意样品中的基质效应,否则,将影响数据的准确性。基质效应的来源:1、内源性物质;2、样品处理过程中引进来的杂质(如tube管的污染);3、样品制备过程中引入的(如增溶剂)消除:1、优化样品前处理;2、优化色谱分离3、更换离子源(通常APCI源的基质效应要小)4、使用同位素内标
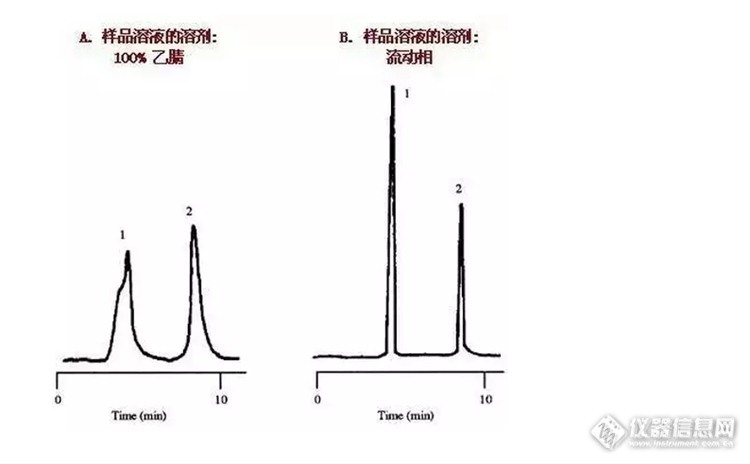
“溶剂效应”会导致哪些峰形异常?如何避免溶剂效应?在HPLC分析中样品溶剂的选择和流动相是怎样的联系?为何建议使用流动相溶解样品?。。。。。一系列的问题都和样品溶剂的选择密切相关,如何解决真正的溶剂效应问题,又如何分辨哪些是非溶剂效应问题,不同的问题有不同的解决方法,有哪些前辈留下的实战经验可供分享呢?一起来看看吧!什么是溶剂效应?样品溶液的溶剂强度强于流动相溶剂强度时可能会造成的峰展宽、峰分叉现象。即色谱图上较早洗脱的峰前沿或者开叉,与此同时较晚洗脱的峰则较为正常的现象。例如样品溶液的溶剂是100%乙腈(100%的强溶剂),而流动相的组成则较弱,18%的乙腈与72%的水。第一个峰是开叉的,并且与第二个峰相比,明显地变宽了。当样品溶液的溶剂变成流动相时,所有的峰形都改善了,且变得尖锐。http://ng1.17img.cn/bbsfiles/images/2016/02/201602251549_585123_2452211_3.png可能发生溶剂效应的情况:出峰时间早;保留弱;进样量大。用流动相溶解样品能够得到很好的峰形 当溶解样品的溶剂不同于流动相时,样品溶剂与流动相发生混合,样品溶剂被冲稀。如果进样溶剂之强度高于流动相,样品在瞬间会表现为在较强溶剂中,并以较快速度通过色谱柱。表现在色谱图上就是∶色谱峰的保留时间缩短。`当进样溶剂与流动相混合时,一部分分子会先与流动相混合,致使这些分子通过色谱柱的速度发生变化,使峰形扭曲,发生变形。洗脱较早的色谱峰峰变形要比洗脱晚的色谱峰严重。解决进样溶剂问题的关键是使进样体积足够小,这样稀释过程会非常快;或使用比流动相弱的溶剂溶解样品。较弱的溶剂会使样品在色谱柱上发生浓缩,在某些情况下,色谱峰会比使用较强溶剂时窄一些。因此,在通常情况下,如果溶解样品的溶剂比流动相强,进样体积应不高于25mL。进样体积大小与进样溶剂与流动相之间的差别大小有关。这一差别很容易凭经验得到∶逐渐加大进样体积,直至发生峰变形现象。采用比发生峰变形时小一些的进样体积即可。问题中发生的峰变形就是因为溶解样品的溶剂甲醇强度远远高于流动相,因而得到了变形的、展宽的峰。如果使用水来溶解样品,溶解样品的溶剂弱于流动相,峰形会得以改善。在离子对色谱中,我们建议使用流动相来溶解样品,以最大程度地降低基线假峰的出现。 样品溶剂分别为100%乙腈、流动相时峰形分析http://ng1.17img.cn/bbsfiles/images/2016/02/201602251550_585124_2452211_3.png上图中,色谱图上较早洗脱的峰扭曲变形或者开叉,与此同时较晚洗脱的峰则较为尖锐与对称,这些现象显示一个比较特殊的起因——样品溶液的溶剂很可能强于流动相。此种强溶剂效应的例子在左图中可见。此处的样品溶液的溶剂是100%乙腈(100%的强溶剂),而流动相的组成则较弱,18%的乙腈与72%的水。第一个峰是开叉的,并且与第二个峰相比,明显地变宽了。当样品溶液的溶剂变成流动相时,所有的峰形都改善了,且变得尖锐。见右图。为什么呢?这是因为当样品进样时,有可能出现峰展宽,最佳的样品溶液组成和体积将会保持在10%甚至更低,在这个例子里,当样品溶剂与流动相溶剂强度不同时,换句话来说,也就是样品未用流动相溶解,因此,有些样品分子溶解在强溶剂中,并随强溶剂流过柱子,而有些则溶解在流动相中,从而导致峰分叉.当样品与流动相强度相差较小,进样影响也会小,第一个峰可能会宽于第二个峰,而当这种展宽导致必要的分离度降低时,这样情况应引起注意,在左图中,使用一根短柱,和5μL进样,这与最佳进样体积4μL相近,用了极性更强的溶剂导致分离度明显的降低,从2.1降到1.5(如右图,分离度为2或更大是评估一个完善方法的一个必要参数,也是每天方法的验证参数,1.5只是一个基本的分离度,任何一个方法或一根柱子都必需满足这个条件,当进样为一倍时,也就是10μL时,分离度更一步降低,此方法就不行了。来源:实验与分析
用溶剂顶替技术能减少沸点歧视效应,改善重复性 这是[url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]中用于不分流中的一种技术,希望得到有关专家解释一下,指点迷津. 还有溶剂聚焦又是什么意思呢?
最近做有机溶剂残留,同时测6种有机溶剂,均稀释溶解在水中,分别为甲醇、乙醇、丙酮、二氯甲烷、三氯甲烷、四氢呋喃。顶空进空白水样,乙醇峰位处有干扰,调整色谱条件,干扰仍无法消除。换用娃哈哈纯净水,也有相似的干扰存在。且峰面积差不多。目前在做这种有机溶剂的检测限和定量限,干扰峰的信噪比将近8,无法测乙醇的信噪比,有想法将空白水样的干扰峰去除,来做定限量。(Agilent 7890带顶空进样器)软件中可以自动扣除空白。不知道是否可以这样做
此贴转自中国色谱网,感谢amian与lishaojing的辛勤工作。-----------------------------------------------------------------------------样品溶剂效应 很多因素可以导致峰形变差。样品溶液的组成与进样体积很可能就是导致此种现象的原因。问题色谱图上较早洗脱的峰扭曲变形或者开叉,与此同时较晚洗脱的峰则较为尖锐与对称,这些现象显示一个比较特殊的起因――样品溶液的溶剂很可能强于流动相。此种强溶剂效应的例子在图10-1A中可见。此处的样品溶液的溶剂是100%乙腈(100%的强溶剂),而流动相的组成则较弱,18%的乙腈与72%的水。第一个峰是开叉的,并且与第二个峰相比,明显地变宽了。当样品溶液的溶剂变成流动相时,所有的峰形都改善了,且变得尖锐。见图10-1B。解释当样品进样时,有可能出现峰展宽,最佳的样品溶液组成和体积将会保持在10%甚至更低,在这个例子里,当样品溶剂与流动相溶剂强度不同时,换句话来说,也就是样品未用流动相溶解,因此,有些样品分子溶解在强溶剂(100%ACE),并随强溶剂流过柱子,而有些则溶解在流动相中,从而导致峰分叉.当样品与流动相强度相差较小,进样影响也会小,第一个峰可能会宽于第二个峰,而当这种展宽导致必要的分离度降低时,这样情况应引起注意,在图10-2A中,使用一根短柱,和5UL进样,这与最佳进样体积4UL相近,用了极性更强的溶剂导致分离度明显的降低,从2.1降到1.5(如图10-2B),分离度为2或更大是评估一个完善方法的一个必要参数,也是每天方法的验证参数,1.5只是一个基本的分离度,任何一个方法或一根柱子都必需满足这个条件,当进样为一倍时,也就是10UL时,分离度更一步降低,此方法就不行了

液相中追求一个高柱效,高分离是我们做色谱追求的目标,但是往往存在很多问题,比如:柱效,分离,拖尾,峰展宽,峰变形,裂缝等等,有些容易解决,有些尝试很多途径都无法改善,当然可能会是方法本身的原因,但是也存在一些忽略的问题:比如本文中提到的溶剂效应,举二例说明:(1)盐酸小檗碱,方法见三黄片补充方法,左图为调整方法前色谱图,峰严重变形,我的做法是调大有机相比例,减小进样量,结果出来右边的图[img=279,149]http://ng1.17img.cn/bbsfiles/images/2010/06/201006111813_223829_1751916_3.jpg[/img][font='Times New Roman'] [img=276,149]http://ng1.17img.cn/bbsfiles/images/2010/06/201006111816_223831_1751916_3.jpg[/img](2)阿莫西林,方法见中国药典二部,左两图为失败方法,右图为调整溶剂的图:需要说明的是我们的是多元泵,也就是作为溶剂的流动相为量筒配置,试验过程中调整了上机的流动相(有机相减少),而作为溶剂的未调。我的做法是将溶剂的流动相调比例至比上机流动相低5个比例。结果见右图。[img=666,149]http://ng1.17img.cn/bbsfiles/images/2010/06/201006111826_223834_1751916_3.jpg[/img]原因,第一例溶剂为甲醇,流动相为乙腈与离子对混合液,第二例溶剂有机相比例比流动相高,共同点为溶剂强度比流动相强。解析:液相一般进样量为10微升,柱子中局部流动相量比较小(没具体算),当样品溶液进入柱子,10微升体积会改变局部的流动相环境(强度),从而可能出现两种保留行为,反应在图谱上就是变形,如拖尾,裂缝,肩峰。希望对坐色谱的朋友有用。[/font]
请问: 什么是溶剂效应、歧化效应?对气相、液相分别有什么影响?
请问用什么软件可以将氢谱中的溶剂峰消除?
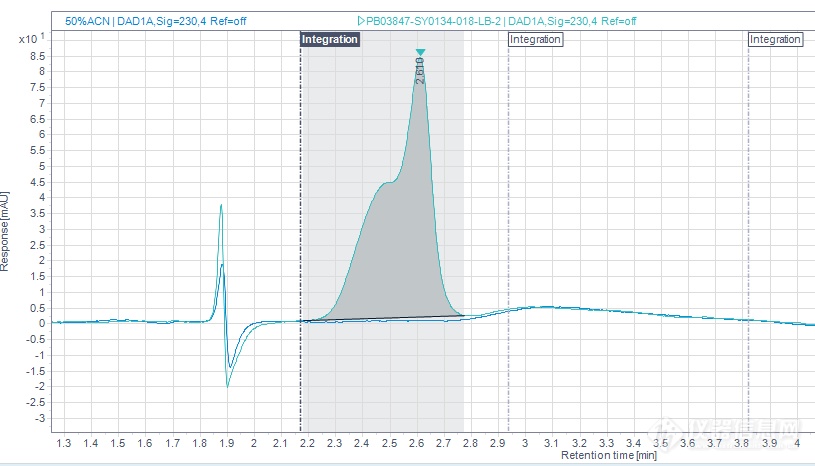
背景在检测某化合物纯度时,样品纯度较低(仅80%),RT=2.6min杂质过大(16.203%),峰形也明显不正常,为肩峰。[img]https://ng1.17img.cn/bbsfiles/images/2022/09/202209011436455003_4200_3433829_3.png[/img]分析方法[img]https://ng1.17img.cn/bbsfiles/images/2022/09/202209011436456820_6668_3433829_3.png[/img]异常调查该样品配样方式:0.5mg/mL,50%ACN稀释,进5ul。对比技术包,发现稀释剂应该用ACN。将样品严格按照技术包( 0.5mg/mL,ACN稀释,进5uL)配制进样,结果发现峰高过载(杂质检测结果失真)。调整进样量为2uL ,重新进样发现RT=2.6min杂质变大,由0.609%→1.947%。[img]https://ng1.17img.cn/bbsfiles/images/2022/09/202209011436459656_2650_3433829_3.png[/img]一开始怀疑是由于样品不稳定造成的,后将样品先配现进( 0.5mg/mL,ACN稀释,进2uL ),并且连续进样,发现样品在ACN中稳定。结果如图:[img]https://ng1.17img.cn/bbsfiles/images/2022/09/202209011436461322_3724_3433829_3.png[/img]杂质裂分成两个明显的峰,且RT=2.4min+RT=2.6min杂质大小保持不变,调取紫外吸收波长显示两者的紫外吸收基本一致。 排除样品不稳定因素,初判断为溶剂效应。异常解决1. 降低进样量 调整进样量(0.5uL 或1uL ),溶剂效应消失。故重新配样( 1.0mg/mL,ACN稀释,进1uL )检测异常样品,RT=2.6min杂质为7.581%。但样品ACN稀释溶解度不好,需超声较长时间。[img]https://ng1.17img.cn/bbsfiles/images/2022/09/202209011436464090_4457_3433829_3.png[/img]2. 更换稀释剂50ACN%稀释进样( 0.5mg/mL,进5uL )溶剂效应存在,进样量为2uL时,溶剂效应消失,但在50%ACN中样品发生水解,RT=2.6min杂质变大(16.037%),用MeOH做稀释剂亦是如此。[img]https://ng1.17img.cn/bbsfiles/images/2022/09/202209011436463753_3884_3433829_3.png[/img]3. 更换与流动相初始比例相近的稀释剂用0.1%TFA:ACN=60:40稀释,进样量5uL,未出现溶剂效应,RT=2.6min杂质为8.189%,样品在该溶剂中稳定存在。[img]https://ng1.17img.cn/bbsfiles/images/2022/09/202209011436464740_222_3433829_3.png[/img]方案3改成余流动相相近的稀释剂适用于该方法的分析总结溶剂效应:[url=https://insevent.instrument.com.cn/t/5p][color=#3333ff]液相[/color][/url]反应中,溶剂的物理和化学性质影响反应平衡和反应速度的效应。可能造成色谱峰展宽、分叉、保留时间漂移、峰面积变化,双峰等现象。与此同时,较早洗脱的峰出现前沿或分叉,较晚洗脱的峰峰形正常。产生原因:样品进入高效[url=https://insevent.instrument.com.cn/t/5p][color=#3333ff]液相色谱[/color][/url]中,当溶剂与流动相存在差异时,一部分样品溶解进入了流动相,一部分还留在溶剂里造成色谱保留的差异。造成这种差异的原因主要有溶剂强度、进样体积、流动相与溶剂的兼容性、溶剂pH等。进样体积导致的溶剂效应:使用强度大于该样品出峰时流动相强度的溶剂,增加进样体积后,目标分析物色谱峰会向前迁移,直至裂分,并且峰高变小峰宽变大。这是因为进样体积较小时,扩散至流动相的目标分析物占绝大多数,除了保留时间略微提前,峰形以及峰宽与流动相溶解时基本一致。体积增大到一定程度,扩散至流动相氛围的目标物与留在溶剂氛围的目标物在数量上相当时,色谱峰分叉明显。当样品体积进一步增大,绝大多数目标物进入色谱柱时是溶解在溶剂中的,目标分析物的色谱峰已经明显向前迁移了。[img]https://ng1.17img.cn/bbsfiles/images/2022/09/202209011436465775_7323_3433829_3.png[/img]PS:当出现样品与空白峰叠不上的情况,可以多注意一下,及时发现异常情况。[img]https://ng1.17img.cn/bbsfiles/images/2022/09/202209011436466927_5663_3433829_3.png[/img]知识拓展溶剂效应产生过程解析:第一阶段:样品刚被注入系统,进入色谱柱后,流动相氛围中的目标物以正常速度被洗脱。因强溶剂洗脱,移动速度快。样品在空间上开始慢慢分裂[img]https://ng1.17img.cn/bbsfiles/images/2022/09/202209011436469431_7193_3433829_3.png[/img]第二阶段:在一定时间内,溶剂氛围中的目标物移动始终快一些,前后两部分距离扩大。但移动快的目标物所处氛围逐渐被流动相稀释(注:因为溶剂和流动相均不保留),其移动速度会逐渐降至正常流速。直到前面目标物的氛围完全被流动相替代,前后两部分的距离稳定下来。因而,样品溶剂与流动相洗脱强度差距越大,两部分目标物最终的距离拉的越大。[img]https://ng1.17img.cn/bbsfiles/images/2022/09/202209011436469085_9601_3433829_3.png[/img] 第三阶段:所有目标物在色谱柱中正常移动,但分裂已不可逆转[img]https://ng1.17img.cn/bbsfiles/images/2022/09/202209011436470189_4645_3433829_3.png[/img]经过检测器,同一目标物给出了两个色谱峰。其中进柱前即扩散进入流动相的那部分目标物组成了后面的色谱峰,它的保留时间与目标物的真实保留时间基本吻合。 更多精彩内容可关注“研发分析之路”,留言小编交流







 我要推广仪器
我要推广仪器
 下载APP
下载APP