原子荧光光谱法测定食品添加剂中砷元素
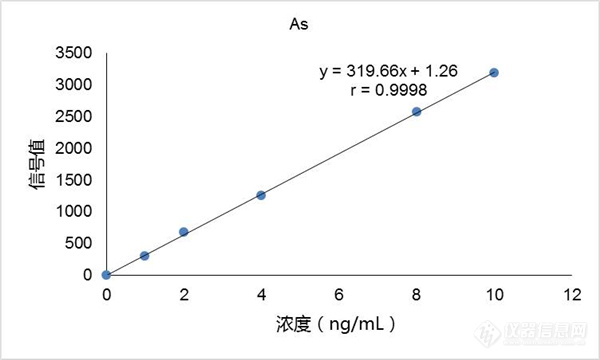
GB 5009.76-2014 食品安全国家标准 食品添加剂中砷的测定代替GB/T 5009.76-2003 食品添加剂中砷的测定,将于2016年3月1日正式实施。标准中将原子荧光光谱法作为食品添加剂中砷的测定方法之一。原子荧光作为检测砷、汞、铅等重金属的常规分析仪器具有灵敏度高、操作简便等特点,而作为中国氢化法原子荧光技术发源地的北京金索坤推出的新一代原子荧光光度计更是具有“多、快、好、省”四大特色。下面为各位实验室检测同行分享下如何应用原子荧光光度计测试食品添加剂中的砷元素。 按照新标准,应用原子荧光光度计测试食品添加剂中的砷元素需要准备以下试剂:氢氧化钠(NaOH)(优级纯)、硼氢化钠或硼氢化钾(NaBH4或KBH4)、硫脲(CH4N2S)、硝酸(HNO3)(优级纯)、硫酸(H2SO4)(优级纯)、高氯酸(HCIO4)(优级纯)、盐酸(HCl)(优级纯)、硝酸镁[Mg(N03)2.6H2O]、氧化镁(MgO)、过氧化氢(H2O2)。 试剂的配制1、氢氧化钠溶液(2 g/L):称取2.0 g氢氧化钠,溶于1 000 mL水中,混匀。2、硼氢化钠溶液(10 g/L):称取10.0 g硼氢化钠,溶于1 000 mL氢氧化钠溶液中,混匀。临用现配(也可称取14 g硼氢化钾代替硼氢化钠)。3、硫脲溶液(50 g/L):称取50 g硫脲,溶于1 000 mL水中,混匀。4、硫酸溶液(1+9):量取100 mL硫酸,小心倒入水900 ml。中,混匀。5、氢氧化钠溶液(100 g/L):称取1.0 g氢氧化钠,溶于10 mL水中。6、盐酸溶液(1+1):量取100 mL盐酸缓慢倒入100 mL水中,混匀,冷却后使用。7、硝酸镁溶液(150 g/L):称取150 g硝酸镁,溶于1 000 mL水中,混匀。 标准溶液的配制1、砷标准储备液(0.1 mg/mL。):精确称取于100℃干燥2h以上的三氧化二砷0.1320 g,加100 g/L氢氧化钠溶液10 mL溶解,用水定量转入1 000 mL容量瓶中,加硫酸溶液(1+9)25 mL定容至刻度。2、砷标准使用液(1/μg/mL):吸取1.00 mL砷储备标准液于100 mL容量瓶中,用水稀释至刻度。 分析步骤以湿法消解为例称取固体试样1 g~2.5 g(精确至0.001 g),液体试样5 g~10 g(精确至0.001 g),置于100 mL锥形瓶中,加硝酸20 mL~40 mL,硫酸1.25 mL,放置过夜。次日置于电热板上加热消解(220℃)。若消解液处理至10 mL左右时仍有未分解物质或色泽变深,取下冷却,补加硝酸5 mL~10 mL,再消解至10 mL左右观察,如此反复两三次,注意避免炭化。如仍不能消解完全,则加入高氯酸1 mL~2 mL,继续加热至消解完全后,再持续加热至高氯酸的白烟散尽,硫酸的白烟开始冒出。取下冷却,加水25 mL,再加热至产生硫酸白烟。取下冷却,用水将消化液转入25 mL容量瓶或比色管中,加入硫脲溶液(50 g/L)2.5 mL,最后用水定容至刻度并混匀,备测。同时做空白试验。 标准系列溶液制备 在25 mL容量瓶中依次准确加入1μg/mL砷标准使用液0 mL、0.05 mL、0.20 mL、0.50 mL、1.00 mL、2.00 mL,硫酸溶液(1+9)12.5 mL,50 g/L硫脲2.5 mL,加水定容至刻度,分别相当于砷浓度0 ng/mL、2 ng/mL、8 ng/mL、20 ng/mL、40 ng/mL、80 ng/mL,混匀备测。 仪器参考条件(以下条件以SK-乐析原子荧光光度计为例)光源:空芯阴极灯,灯电流60~80mA 负高压:-300~-350V 主气流量:为定值,500mL/min左右 辅气流量:800~1000mL/min泵速:70~80转/min检出限(参考值):0.01ng/mL 注意事项:(1)在盐酸中一般都存在着一定含量的As,因此采用优级纯HCL可减少空白。但也有个别情况分析纯中As含量低于优级纯,以及不同生产厂或不同的生产批号As的含量差别也很大, 因此建议在使用前先用少量的HCl配制成10%(V/V)条件下进行对比检验。(2)将所使用前的各种器皿必须用(1+1)HNO3浸泡24小时,然后认真清洗干净,防止As的污染。(3)本说明所配制的砷标准贮备液为三价状态,为防止在保存期间砷被氧化,仍建议加入硫脲+抗坏血酸,碘化钾预先还原As(Ⅴ)至As(Ⅲ),还原速度受温度影响,室温低于或小于15℃,至少应放置30分钟,样品也必须同样进行预还原。(4)配置标准溶液的容量瓶必须长期固定不变,不能任意变动。(5)配制标准溶液时宜采用固定的一支5mL刻度的移液管,可直接用于配制全部标准系列。(6)硼氢化钾溶液浓度对As测定有较大影响。






























 我要推广仪器
我要推广仪器
 下载APP
下载APP