想请教下,质谱和色谱定量的差别?
请问各位专家,用[url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]-质谱分析农药残留做定性定量分析时,怎么选择定性和定量离子?依据是什么?谁能分享一下相关的资料!谢谢!
质谱的应用越来越广泛,良好的定性能力是大家公认的,但是说到定量,很多人都认为质谱的定量结果准确性不如色谱,真的是这样吗?发表一下您的看法吧。好的回帖另有加分,技术版面,请勿灌水!
我之前定量没有用同位素内标,保留时间也能分开。最近看到关于质谱定量内标选择的内容,认为同位素内标好,能校正基质效应,色谱行为和响应特征接近,即便没有合适的同位素内标,也要选择结构类似的,色谱行为接近的。但是如果液相分不开一起进质谱的话,会不会产生离子抑制?
高效液相色谱串联质谱可以定量吗?
请问您在没有质谱情况下,用过双色谱柱定性定量吗?
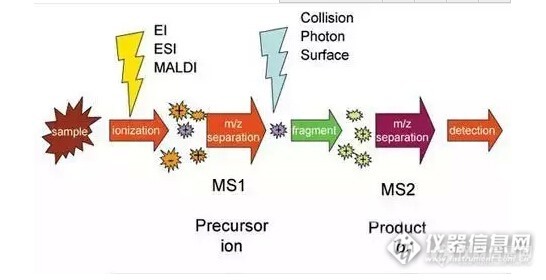
质谱信号。与EI谱图分析以相对强度为主不同,在色谱-质谱联用时,信号的绝对强度就成了我们天天都要关心的内容,因为质谱信号强度随时间的变化就是实验的色谱图,通常以总离子强度或者某一特定质荷比离子的强度作图。http://ng1.17img.cn/bbsfiles/images/2015/11/201511271813_575350_2544766_3.jpg2、定量的两种方法外标法 用已知量的标准样品A和未知量的待测样品A分别进行实验;我们会得到以下三个信息:标准样品的量(已知);标准样品的信号强度;待测样品的信号强度。(假设样品的响应=常数*浓度,从这三个信息即可算出待测样品的量。) 为了更加精确地测定未知量的样品,我们希望标准样品的信号强度与待测样品的信号强度尽量接近(以减少非线性响应的影响)。因此常用的外标法会测量一系列已知量的标准样品,绘制一条工作曲线,再用拟合的方法确定未知样的量。http://ng1.17img.cn/bbsfiles/images/2015/11/201511271814_575351_2544766_3.jpg内标法 外标法主要有以下两方面的局限:1标样和待测样是独立进行实验的,实验间的偶然误差无法消除;2标样和待测样的基质(即除待分析物外的其它成分)不同,基质有可能会带来不同的影响,也会产生误差。 那么,如果我们把已知量的标准样品B直接加入待测样品A,就可以把标准样品和未知样品的测定在同一次实验和同样基质中完成,也就消除了两次实验和基质不同造成的误差,这就是内标法。(如果加入的标准样品和待测样品是同种物质A,那么由于它们不可区分,只通过一次实验是不能定量待测样的,这时我们在加入标样前后分别进行两次测量,即测量待测样及待测样+标样的信号,即可计算出待测样的量。)3、质谱相关的特殊定量细节同位素稀释 前面内标法的介绍中我们可以发现,最理想的内标物既要和待测样相同(具有相同的响应系数)又要不同(仪器可以区分二者的信号),这对矛盾的集合体就是同位素内标。 由于不同同位素的化合物具有近似相同的物理化学性质,离子化时的响应通常也是相同的,而它们具有不同的质荷比m/z,即可在质谱中被区分出来。因此同位素标准品是最理想的内标物。 另外,由于某些元素的天然同位素分布有一定的比例,当我们加入一定量的同位素内标时,可以把对信号绝对强度的测量转化为对信号相对比例的测量,从而提高实验的准确性。http://ng1.17img.cn/bbsfiles/images/2015/11/201511271814_575353_2544766_3.jpg选择反应监测 在不太复杂的体系中,我们只要按照分子量就可以定性某种化合物了。但对于复杂混合物(如石油产品/生物样品)而言,很多化合物具有相同或相近的质量(同分异构体质量完全相同,有些化合物分子量非常接近,如CO和N2,要考虑仪器的质量分辨率是否能区分二者),此时仅靠测量质量就不能确定这个化合物是否就是我们关心的“the one”了。 在串联质谱 (Tandem MS) 仪器中,我们不仅可以把质谱仪理解为一个称量离子的“天平”,它还具有了离子“镊子”(选择某个特定的离子把它分离出来)和“剪刀”(把某个/某些离子激活并打成碎片)的功能。通过母离子和子离子的两步选择,我们可以在复杂体系中精确定位到我们关心的化合物,同时,两次离子选择还可减少复杂基质的干扰,降低背景噪声(获得更低的检出限)并提高方法的动态范围。因此选择反应监测是目前色谱(气相色谱/液相色谱)-质谱联用中最常用的定量方法。http://ng1.17img.cn/bbsfiles/images/2015/11/201511271815_575354_2544766_3.jpg选择反应监测在不太复杂的体系中,我们只要按照分子量就可以定性某种化合物了。但对于复杂混合物(如石油产品/生物样品)而言,很多化合物具有相同或相近的质量(同分异构体质量完全相同,有些化合物分子量非常接近,如CO和N2,要考虑仪器的质量分辨率是否能区分二者),此时仅靠测量质量就不能确定这个化合物是否就是我们关心的“the one”了。在串联质谱 (Tandem MS) 仪器中,我们不仅可以把质谱仪理解为一个称量离子的“天平”,它还具有了离子“镊子”(选择某个特定的离子把它分离出来)和“剪刀”(把某个/某些离子激活并打成碎片)的功能。通过母离子和子离子的两步选择,我们可以在复杂体系中精确定位到我们关心的化合物,同时,两次离子选择还可减少复杂基质的干扰,降低背景噪声(获得更低的检出限)并提高方法的动态范围。因此选择反应监测是目前色谱(气相色谱/液相色谱)-质谱联用中最常用的定量方法。
顶空固相微萃取[url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]质谱联用测定挥发性物质 如何定量?
是这样的,质谱例如乙苯的定量离子是91/106,那么我看质谱图的时候发现它的碎片是91.1/106.1,再进一针发现它的碎片是91/106,在做线的时候选择的定量离子是91.1/106.1,这样是对于91/106是可以正常定量的吗,会对结果有什么影响吗,例如定不了量之类的,感谢老师??
关于质谱图中的定量离子与定性离子的确定方法:定量离子是不是以质谱图中最高的相对离子强度为定量离子,定性离子的选择则以特征离子的丰度比为选择依据啊,我是初学者,多谢指教
我用的是热电的GC-MS,对于软件的操作不是很熟悉。由于我是刚接触对于质谱的定量问题一直不是很清楚,看了一些书,介绍的方法和单纯气谱的差不多。所以,想请各位高手指点一下,如何利用质谱图进行定量分析呀!谢谢各位的帮忙!
1 要用目标离子的碎片定量,特征性强,排除干扰;2 在定量分析的方法设置上,尽可能提高扫描速率,提高准确率和重复性(可以通过a减小扫描质量数的范围来提高目标峰的扫描次数,或 将一个样品全部分析时间断分成n个segments,对目标离子单独设置扫描模式);3 一定要通过色谱柱分离后定量分析,避免竞争性离子的存在影响目标离子的离子化效率;如果目标分子未与竞争性分子完全分开,则在离子化过程中导致目标分子的离子化效率降低,导致样品分子的定量结果偏低,当然标准浓度的样品也要用相同的方法分析。4 如果样品都是纯品的话可以不经过色谱柱直接进样分析,包括做标准曲线的样品(虽然不建议直接进样分析)。5 如果用的离子源的喷针位置是可移的话,一定要记住做标准曲线时其位置,否则其位置移动后在相同的条件下进入质谱的离子流量会发生改变,标准曲线就不能使用了,白忙!对于调用的质谱方法不要改动shealth gas and aux gas 的流速,否则会影响进入质谱的样品量(谢谢Esquire提醒) 6 所建立的标准曲线一个月后如果想重复使用,则用QC样品检验一下该标准曲线!7 对于已经建立好的分析方法在扫描范围、流动相的组成、梯度或流速等方面不要作任何改动,否则,标准曲线要重作。扫描范围改变目标峰的扫描次数、流动相组成改变离子化效率,流速改变色谱峰的保留时间和峰宽。8 离子阱的强项在于多级-定性,四级杆的强项在于定量;9 对于热稳定性不好的样品可以通过提高气速,降低毛细管温度的方法保证定量分析的重复性;一旦方法固定后不要轻易改动;10 仅供参考,欢迎探讨!!
如我要测试的混合气体中包含CH4、H2和N2,怎样用质谱对其进行定量呢?我们的是一台普通的四极杆质谱,不带色谱。谢谢!
各位老师,大家好,新学质谱定量分析,请问质谱定量要用哪一种模式呢?我们做的一级质谱的信噪比很低啊,还不及紫外,定量为什么要用到二级质谱呢?烦请各位老师赐教。
请教各位专家:我用的是四级质谱,想根据特征峰用它进行定量。但是我发现在同一条件下,纯物质的各个碎片峰比例不是定值,比如说噻吩,它的两个比较大的峰是m/z84和58,可是测定的过程中,两个峰的强度比值在不同的时间不相同,请问这是什么原因啊?如果比值不定我就无法用特征峰进行定量。多谢
[color=#444444]质谱是四极杆检测器。请问其定量方法是怎样的?[url=https://insevent.instrument.com.cn/t/bp][color=#3333ff]气质联用[/color][/url]时[url=https://insevent.instrument.com.cn/t/Mp]气相[/url]定的是峰面积,那么质谱定量定的是什么?怎么定的。求详解[/color]
质谱的定量效果如何?
我用质谱做样时,最后定量出来匹配度很低,我把时间范围度小点匹配度就高了,是怎么回事?
三重四级杆质谱是如何定量的?三重四级杆通过离子打碎获得特异性子离子,子离子在通过Q3后时在接收器上转化为电信号,反映到分析软件就是离子强度图。在一定线性范围下,分析物浓度越高,打到接收器上的离子就越多,信号越强,这是定量的基础。此外在定量时可以选择用峰高或峰面积定量,一般选用峰面积定量准确。因为是定量,所以必需标准曲线,原理与高效[url=https://insevent.instrument.com.cn/t/5p][color=#3333ff]液相[/color][/url]类似。一般多用内标法定量,以排除质谱重现性和样品处理造成的影响。那么,随着设置离子对越多,如何能保证定量准确呢?四极杆对于离子的选择性通过是交替进行的,在所有离子通道之间快速切换。随着设置离子对数量的增加,每个离子所占用的检测时间缩短,这会影响到其检测灵敏度,但是只要实际样品和标准溶液所采用的分析方法一致,定量的准确性理论上是不受影响的。
各位高手:我刚开始做质谱,请多指教。 我的质谱是5973,我知cas号或进标样,怎么查看这性和定量离子呀。最好是举例说明,在此先谢了。
分析样品是多糖的酶解产物,LC-MS得到TIC图,由于没有标准品也没有标准质谱图,所以只能在TIC中每个峰尖处进行定性,请教各位,是否能用TIC每个峰的峰面积进行相对定量?另外,SIM定量具体指什么?我的TIC里有好几个峰,怎么用特征离子定量,是不是有几个峰就重复进几次样,选择不同的特征离子。
【网络讲堂】:基于Orbitrap质谱的定量蛋白质组学技术新进展【讲座时间】:2015年01月29日 14:00【主讲人】:李静 (赛默飞世尔公司色谱质谱部应用研究中心质谱应用工程师,在赛默飞一直致力于蛋白质组学的技术支持,积累有丰富的分析经验。)【会议简介】1、 2015 CNHUPO生物质谱蛋白质定量高级研讨会新进展介绍2、 Thermo DIA(数据非依赖采集)定量技术新应用3、 Thermo TMT SPS MS3定量技术新应用-------------------------------------------------------------------------------1、报名条件:只要您是仪器网注册用户均可报名参加。2、报名并参会用户有机会获得100元手机充值卡一张哦~3、报名截止时间:2015年01月29日 13:304、报名参会: http://www.instrument.com.cn/webinar/meeting/meetingInsidePage/13225、报名及参会咨询:QQ群—231246773
有没有人做过质谱绝对定量蛋白的?
你好,如果我采用质谱定量,用内标法,内标的含量保持恒定,按比例增加标准样品的浓度,以标准样品和内标的比值及标准样品的浓度做标准曲线,会不会因为标样的浓度越来越大,产生离子抑制,而使标准曲线趋势降低,结果偏低?
大家先看看这段话,合理吗?(1) DAD数据处理系统可为用户提供各种类型的色谱图,其中包括:单波长色谱图、任意两个波长的吸收比色谱图、波长时间程序色谱图、最大吸收波长色谱图以及总体吸收色谱图。其中最大吸收波长色谱图为灵敏度最高的检测方式,而总体吸收色谱图为定量重复性最好的方法。有人看到“总体吸收色谱图为定量重复性最好的方法”这句话,就打算在实际操作中使用......我晕......我可从来没有看到哪个HPLC用这个所谓的总体吸收色谱图定量的。质谱里才用用TAC,还不是推荐选项......
Introduction to MS Quantitation and Modes of LC/MS Monitoring[img]http://www.instrument.com.cn/bbs/images/affix.gif[/img][url=http://www.instrument.com.cn/bbs/download.asp?ID=15863]质谱定量及定量模式介绍[/url]
谁能提供一些有关质谱定量分析的经验?谢谢!!
如题,用选择性离子扫描时,质谱的定性和定量峰是如何选择的?分子离子峰可用做什么?谢谢,新手求助!
[align=center]API质谱仪MRM定量方法的建立方法[/align] 质谱仪凭借其高灵敏度和选择性,定量小分子化合物越来越受到重视。MRM:Multi Reaction Monitor,指多反应监测。针对二级质谱或多级质谱的某两级之间,即母离子选一个离子,碰撞后,从形成的子离子中也只选一个离子。因为两次都只选单离子,所以噪音和干扰被排除得更多,灵敏度信噪比会更高,尤其对于复杂的、基质背景高的样品。那么我们今天来分享一下API质谱仪MRM定量方法的建立方法。 首先选择Q1 FULLSCAN:用SYRINGE PUMP 5ul/min,样品浓度约1-10pmol/u,根据待测化合物性质选择+- ESI/APCI/APPI。根据待测化合物分子量选择扫描范围:在分子量上下各100Da足够,TIME不要小于1秒。通过调节DP,使得分子离子峰明显高于噪音峰,确定分子离子。如果分子离子峰不明显,可能需要增加样品浓度,如果仍然不能确定分子离子,考虑改变离子化方式或样品前处理方法,例如POS方式可酸化溶液,NEG方式碱化溶液。对于某些化合物,POS方式[sup]+[/sup]不明显,可考虑[M+NH[sub]4[/sub]][sup]+[/sup],尽量避免采用[sup]+[/sup],[sup]+[/sup]。 第二步选择Q1 MI SCAN:TIME 100ms,用SYRINGEPUMP 5ul/min,根据第1步选择的母离子,EDIT RAMP优化DP,CXP及EP,存储此参数。 第三步选择PRODUCT ION SCAN,用SYRINGE PUMP5ul/min,扫描范围:上限比母离子高10-50 Da,下限50-100 Da,TIME 不小于1s。通过调节CE等参数得到高质量的MS2图,母离子相对峰高在1/4-1/2即可。Acquire命名 MCA20次,平滑后选择特征子离子。 第四步选择MRM优化,用SYRINGE PUMP5ul/min。根据特征子离子强度选择MRM离子对,母离子M/Z值由第1步确定,子离子M/Z值由第3步确定,精确到小数点后1位。可选择多个MRM离子对,同时优化,对较弱离子对可适当多分配TIME例如200 ms,强离子对可40-50 ms。同时EDIT RAMP优化COMPOUND项下参数先根据第3步的CE优化CXP,然后再优化CE,在质量扫描范围窗口点右键对各离子参数分别设定。CAD参数可手动优化,值不要太高,通常4-6。初步建立METHOD,存储为*.dam文件。 第五步不接色谱柱进行FIA定量优化。首先接通LC系统,检查工作是否正常,有无气泡,否则先排气。激活CONFIGURE中[url=https://insevent.instrument.com.cn/t/Yp][color=#3333ff]LC-MS[/color][/url]配置。在ACQURE栏内调出第3步初步建立的METHOD,若已经有LC条件,则可参考,若无根据拟选用的色谱柱内径确定LC流速(2mm典型流速200-300ul/min,3mm典型流速400-500ul/min,4.6 mm典型流速800-1000ul/min)以及流动相组成,设定MS初始温度和气流,加上LC PUMP和AUTOSAMPLER,并设定同步方式为LC SYNC。 第六步使用4mm以上内径色谱柱时先设定分流比,使进入MS的流量在200-400ul/min最佳。MS与LC的period都设为0.5-1min,进样体积2-5ul,浓度0.01-0.1pmol/ul,按照FIA教程操作,优化CUR,GAS1,GAS2(API3000AUX要手动优化),TEMP,IS。平衡5 min后开始进行FIA定量优化,完成最后方法的质谱条件优化。 第七步接好并平衡色谱柱,至少20min,用适当浓度标准品检验峰型等分离情况。根据色谱分离情况,可设定梯度洗脱。MRM可以不必所有峰都基线分离,但要避开基质的离子抑制干扰(出峰时间不能太早,要在溶剂峰后)。 第八步空白基质添加标准品,稀释成不同浓度,通常可2倍等比稀释,5-7级。根据LC流动相选择溶剂体系一致的稀释液,配制实际样品。编辑批处理文件,排列进样顺序,最好第一针先做空白,然后由稀到浓。平衡MS离子源和色谱柱后开始正式采样。 以上是一种化合物的定量方法,如果多种物质同时定量(包括内标),分别用纯标样重复第2-4步,记录各参数,然后合并到第5步最后的METHOD,再进行第6-8步。完成上述所有步骤后,即可制做标准曲线。如果不能自动正确积分色谱图,可手工修改。有内标和无内标的计算方法分别演示。最后结果的打印和存贮,复制。 今天的分享到此结束,感谢仪器信息网提供原创大赛让我们有机会互相分享学习!
需要定量分析CH4,CO2,CO,H2,反应是CH4+CO2=2CO+2H2,正在想法对其进行标定,得出反应的转化率。使用CH4,CO2,CO,H2纯气,按比例混合后进入质谱进行分析,如何进行试验数据处理,如何进行试验能使标定误差最小?求高人指点







 我要推广仪器
我要推广仪器
 下载APP
下载APP