我的离子色谱世界(下篇)——离子色谱的“七子之歌”
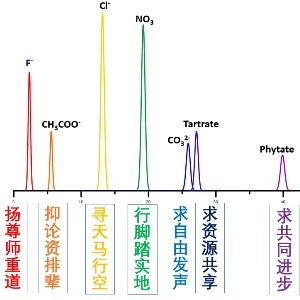
p span style="color: rgb(0, 112, 192) "strong前言:/strong/span七子之歌,闻一多1925留美期间创作,共七首,分别是澳门、香港、台湾、威海卫、广州湾、九龙、旅大(旅顺-大连)有着特别的历史背景。今引用于此,主要取意于其在艰苦环境中的一种美好的盼望,以下也是我对离子色谱的美好盼望与期待。其实,看图片摘要就已经一目了然(英文期刊Graphical abstract 很重要),柱子做的还是炒饭。一个人的看见是有限的,但是我们每个人的看见分享出来,大家就能一起看到整个图片。那就先看我怎么讲一个重复而老旧的故事吧(新瓶装旧酒)!/pp style="text-align: center "img src="http://img1.17img.cn/17img/images/201803/insimg/3e2a8842-3bb2-47e6-b0f0-ff7c636f4454.jpg" title="七子之歌图片摘要2.jpg" width="450" height="417" border="0" hspace="0" vspace="0" style="width: 450px height: 417px "//pp span style="color: rgb(0, 112, 192) "strong故事背景/strong/span(离子色谱各部件的比喻):/pp span style="color: rgb(0, 112, 192) "strong泵前纯水源:/strong/span汽车车轮,有它才可能有下面的故事,没有它就什么都不会发生;/pp span style="color: rgb(0, 112, 192) "strong泵头:/strong/span汽车引擎,没有它,一切都将沉寂如死水;/pp span style="color: rgb(0, 112, 192) "strong淋洗液发生器:/strong/span汽车油门,车能跑多快就全靠加多大的油门。如果跑得太快,就分不清人、事、物,会错过许多美景;/pp span style="color: rgb(0, 112, 192) "strong分离柱:/strong/span犹如三棱镜一样,将白光分成绚丽的彩虹。又好比从中药药方中提取出一个个有效成分,用来治愈疾病。今天我们将提炼出七个主要的有效成分;/pp span style="color: rgb(0, 112, 192) "strong抑制器:/strong/span抑制住中国传统文化中的糟粕部分,也就是除去巨大的噪音背景,从而水落石出;/pp span style="color: rgb(0, 112, 192) "strong电导检测器:/strong/span显扬这个时代所需要的正能量,宣扬积极与正面的理念(比方说“为你点赞”);/pp span style="color: rgb(0, 112, 192) "strong七子之歌的故事/strong/span/pp 用图片摘要中的七个离子来代表洪柱所追求的离子色谱七小点,且看我如何娓娓道来。/pp 前四个离子是一价的,代表着我们个人所需要做到和追求的,就好比是建筑的根基,没有它们四者就没有后面的三者!/pp 紧接的是两个二价离子,代表着我们集体的追求,就是“求自由发声”与“求资源共享”!/pp 最后一个是十二价离子,代表着我们集体最高的理想与追求,那就是“求共同进步”!/pp span style="color: rgb(0, 112, 192) "strong下面就听我唱一曲《七子之歌》吧!/strong/span/pp span style="color: rgb(0, 112, 192) "strongFsup-/sup:/strong/spanFsup-/sup是第一个洗涤出来的离子(暂且忽略其前面可能有其它不常见离子)。 “尊师重道”是我们传统文化所宣扬的美德,也应当是我们所需要看重的第一样个人品质,,在此就不展开叙述。就说两个不常见的,那就是F-有两个功用,一个是防腐,一个是发光。前者是含氟牙膏,后者是夜明珠。“扬尊师重道”,真正的尊重技术与人才,尊重行业前辈,这个行来就永远不会腐朽,而是会不断发光。所以,扬“尊师重道”!/pp strong span style="color: rgb(0, 112, 192) "CHsub3/subCOOsup-/sup:/span/strong是一个有机弱酸,出峰时间一般在Fsup-/sup之后。正如它通常不完全电离的性质一样,CHsub3/subCOOsup-/sup(意指论资排辈)是当受抑制的成分。而CHsub3/subCOOH正如其名,因为它就是醋的本身与实质。有关“吃醋”来源的唐朝故事,大家应当都听说过吧,就不多言了。同行相妒,同行相轻就是这醋的缘故。经过时间的发酵,它就变为了陈醋,就是中国人心里几千年来的阶级制度,有出身高低贵贱之分,有论资排辈之嫌。“抑论资排辈”就是要抑制这醋,它也是这“七子”之中唯一消极的。只有抑制住,才能实现人与人之间的平等与尊重,因为刚入行的,入行十几年乃至几十年的,心中都有他自己的离子色谱世界。/pp span style="color: rgb(0, 112, 192) "strongClsup-/sup:/strong /spanClsup-/sup是飘浮不定的,来无影去无踪。它可以来自于空气,来自于样品,来自于水源,来自于各种化学器皿。总之,在离子色谱的谱图上或多或少总能见着它的身影。这Clsup-/sup就是我们里面的想象力。我们看重它,它就会长大;我们轻视它,它就会减少。有人讲Clsup-/sup这个东西太虚化,我们要干实事不需要。可是,回头想一想,我们每一天吃了多少的Clsup-/sup,就不要责备它了。Clsup-/sup吃多了,我们多喝几口水,还是可以接着做实事,不耽搁。反过来,我们的脑袋无时无刻不在想象漂浮之中,或在这里或在那里。再想象一下,没有盐的生活将是多么的枯燥无味。“寻天马行空”就是去除中国人“师承”的思想禁锢(“师承”本意即学生的思想与理念,发表的作品与观点不能超过导师所画的框架。)这样,才能有学术思想自由的天空。/pp span style="color: rgb(0, 112, 192) " strongNOsub3/subsup-/sup:/strong /spanNOsub3/subsup-/sup它是非常踏实的一位,也见于我们所接触的多数水体样品之中。“脚踏实地”的NOsub3/sub-,不仅电导检测器能看见它,紫外可见检测器也看得见它。它多多少少总存在于我们中国人的美德之中,正是这美德,让海外的中国人在科研学术中作出了卓越的贡献。各行各业,古今中外华夏儿女有很多这样的杰出之士,就不一一列举了。想提到的就是NOsub3/subsup-/sup很容易就变成了NOsub2/subsup-/sup,那就是“脚踏实地”的两个反义词,一个是“偷工减料”,一个是“好高骛远”,这二者都是极为有毒的成分,也给中国人“脚踏实地”的美德蒙灰而褪色。今天,我们就一起努力,再加回这个O原子,让NOsub3/subsup-/sup增多,让NOsub2/subsup-/sup减少。另外,NOsub3/subsup-/sup虽然没有Clsup-/sup峰高,可是它的面积更大,所以意义更重要。“行脚踏实地”就是学习德国人、日本人追求极致的态度与理念,推崇匠人文化,十年磨一剑(国内虽然整体没有这样的风气,但是我已经遇到好多这样的例子了),寻求超越前人。/pp span style="color: rgb(0, 112, 192) " strongCOsub3/subsup2-/sup与tartrate/strong:/span这二者都是二价离子,在离子色谱上相隔很近,有时很难分开,所以一起来讲。前者对应着“自由发声”,后者对应着“资源共享”。COsub2/sub无处不在,虽然它在现代的离子色谱的分离中常常是一种干扰,干扰其它离子的定量检测。可是它在经典传统的离子色谱中,却是一个“好人”,成就了其它离子的分离与测定,而自己消失在背景之中,是个无私的角色。COsub3/subsup2-/sup就如鱼塘里青蛙的叫声,或多或少,你总能听见。有人看是好的,有人看是不好的,全在于你的角度。Tartrate(酒石酸根)发现于1769年,关于它被发现的故事很有意思,感兴趣的可以自己去查找一下,这里也正是取意于此。只有资源共享,我们才不会浪费时间与精力去重复别人已经做过的工作,才会更快地发现新的东西。站在前人的肩膀上,我们才能看得更远。让我们自由分享已经发表的工作,分享或失败或成功的实验经历,分享离子色谱新的技术与产品,分享离子色谱背后有趣的故事,让我们在一个更好的舞台上共同起舞。自由发声才能激发人的思维发散,资源共享才是脚踏实地向前的推动力;自由发声才能实现人与人之间的平等相待,资源共享才能打破壁垒,实现科学无国界。/pp span style="color: rgb(0, 112, 192) "strongPhytate(肌醇六磷酸):/strong/spanstrong /strong本来想选citrate(柠檬酸盐),后来发现Phytate更好,有更好的象征意义。同时,它涉及到我已经发表的两篇文章(请补充文章链接)[1,2],那就算是广告植入吧。Phytate (意指共同进步)是我们的终极梦想。Phytate做为一个带12价的阴离子,按理讲它永远不会有出头之日。它会被分离柱死死地抓住,因为梦想与现实正负差异太大了。就算将淋洗液发生器的油门踩到最大时,按理讲也无法砍断它12道防锁,总是藕断丝连。梦想终究逃脱不了现实的残酷禁锢,只能做为梦想而已。可是,它最终神奇地出现了,跑到了马拉松赛的终点,电导检测器记录下它的到达时间,39分59秒(取意于其在我所用色谱柱上的出峰时间约为40 分钟)。梦想庞大的身躯使它不留恋于环境的拦阻(空间位阻效应)。路边不断加油的拉拉队给了它前进的动力(淋洗液中的阳离子成分能减少Phytate实际的有效负电荷),这两个神助攻,加速了它的前进。总之,Phytate能突破柱子里面的重重拦阻,出现在离子色谱的谱图上,那“共同进步”的梦想也会出现在离子色谱世界。span style="color: rgb(0, 112, 192) "strong前面所提的六条的神助攻就能实现共同进步的理念,这是一种双赢的局面,那就让我们一起做个大蛋糕吧!/strong/span/pp 总而然之,这是前面所写《我的离子色谱世界》与《柱子”离谱“的中国梦》的残羹冷饭做成的炒饭。我们中间有很多厉害的人物,肯定有更多可实现有用的理念。柱子只是做青蛙的,下面就来讲讲青蛙。青蛙有几个特点:span style="color: rgb(0, 112, 192) "strong一是眼睛大/strong/span(好像看见东西,实则啥也没看见,只有外面动的肤浅的才看得见);span style="color: rgb(0, 112, 192) "strong二是爱哇哇叫/strong/span(即不能像黄鹂一样有美妙的歌声,也不能像喜鹊一样带来好消息就是呱呱叫);span style="color: rgb(0, 112, 192) "strong三是爱冬眠/strong/span(当群里冷静时它也不发出叫声了);span style="color: rgb(0, 112, 192) "strong四是肚子鼓鼓的/strong/span(看似里面有货,实则是空气无用之物,虚张声势而已)。但是,这又怎么样呢?span style="color: rgb(0, 112, 192) "strong当大鱼被吸引起来时,青蛙就满了意义!/strong/span/pp 【1】 Anion Composition of Acai Extracts/pp a href="https://pubs.acs.org/doi/abs/10.1021/jf4014185" _src="https://pubs.acs.org/doi/abs/10.1021/jf4014185" style="color: rgb(0, 112, 192) text-decoration: underline "span style="color: rgb(0, 112, 192) "https://pubs.acs.org/doi/abs/10.1021/jf4014185/span/a /pp 【2】 Enigmatic Ion-Exchange Behavior of myo-Inositol Phosphatesbr//pp span style="color: rgb(0, 112, 192) " /spana href="https://pubs.acs.org/doi/abs/10.1021/acs.analchem.5b00351" _src="https://pubs.acs.org/doi/abs/10.1021/acs.analchem.5b00351" style="color: rgb(0, 112, 192) text-decoration: underline "span style="color: rgb(0, 112, 192) "https://pubs.acs.org/doi/abs/10.1021/acs.analchem.5b00351/span/a /pp style="text-align: right " 供稿人:廖洪柱博士/pp 德克萨斯大学阿灵顿分校分析化学博士,博士期间主要是借助离子色谱仪与柱后碱引入方法实现对极弱酸的灵敏检测。先后开发出小体积高混合率的在线混合器,挥发性弱酸(硫化氫与氰化氢等)的转移与检测装置,以及挥发性胺的引入装置并申请了相关国际专利。现就职于德克萨斯州NEOS Therapeutics公司,该公司主要开发ADHD(专注力失调与过度活跃症)类缓释药物,主要利用离子交换树脂来吸附与缓释药物有效成分,目前公司已有三款新药上市。作为研发部门的一员,一方面专注于药物分析方法的开发与验证,另一方面专注于新药的研发工作,在离子色谱,高效液相色谱,液质联用,扫描电镜仪等仪器的应用方面有较深入研究。/p


























 我要推广仪器
我要推广仪器
 下载APP
下载APP