同位素稀释质谱法分析痕量钚建立了同位素稀释质谱法分析测定痕量钚的分析技术。应用该技术分析了239Pu丰度为94%的同位素标准样品,当样品量为100pg时,样品中的240Pu/239Pu分析值的不确定度为20%(1s),与传统的钚同位素分析方法相比较,使钚的分析测试能力提高了两个数量级。该分析技术包括以下三个部分: 239Pu丰度标准样品的浓度,采用a绝对测量的方法来测定。结果为:7.227(1±0.015)ng 239Pu/mg溶液;242Pu稀释剂的浓度,用已知浓度和丰度的239Pu来标定,标定结果为:0.1815ng 242Pu /mg溶液;痕量钚的分析测定,用242Pu作稀释剂(10~20ng),加入100,500,1000pg的239Pu丰度为94%的同位素标准样品进行痕量钚的分析,测定标准样品中的240Pu/239Pu比值,并与标称值0.05814进行比较,测定结果见表1。表 1 痕量钚同位素分析结果 样品 239Pu/pg RM92* RM02 RM12 R09 偏差 1 92.4 0.020261(1±0.015) 0.018319(1±0.046) 0.001620(1±0.098) 0.0674(1±0.20) +16% 2 460.4 0.053100(1±0.0027) 0.020448(1±0.035) 0.001517(1±0.011) 0.0659(1±0.27) +13% 3 930.8 0.085967(1±0.0040) 0.02168(1±0.0038) 0.001602(1±0.026) 0.0598(1±0.026) +2.8% 注*:RM92为240Pu与239Pu混合样品中的240Pu/239Pu比值。 从表中数据可以看出:R09不确定度的主要贡献是稀释剂中240Pu与242Pu比值测量的不确定度,准确测量它们的比值是降低痕量钚同位素分析的不确定度的关键。
准备上一台硫化物分析的GC。COS检测标准为20ppb。如此低浓度的标样比较难找,只有想其他办法。查阅资料并咨询厂家后得到两种技术:1、定制较高浓度标气,并选配自动稀释装置获得相应低浓度标气。如安捷伦。2、选用硫渗透管,通过控制冲洗气体流量获得相应低浓度标气。如AC。问题:这两种方法效果如何?可以做到多低浓度的标气?准确性如何?操作上方便么?希望有经验的大侠们伸出援手!谢谢!
高纯气体标准物质中微量或痕量杂质分析的要求有哪些?
有人做过用紫外差分吸收检测痕量气体吗?我对里面的吸收截面不明白,应该怎末理解呢?这个吸收截面是怎末测量的?
最近需要做一气体的痕量杂质元素的检测。不知道通常的收集方式和处理是怎样的?望各位大侠赐教!万分感谢。
地质样品中痕量铼的同位素稀释[url=https://insevent.instrument.com.cn/t/yp][color=#3333ff]ICP-MS[/color][/url]准确测定方法研究[img]http://www.instrument.com.cn/bbs/images/affix.gif[/img][url=http://www.instrument.com.cn/bbs/download.asp?ID=28175]地质样品中痕量铼的同位素稀释[url=https://insevent.instrument.com.cn/t/yp][color=#3333ff]ICP-MS[/color][/url][/url]
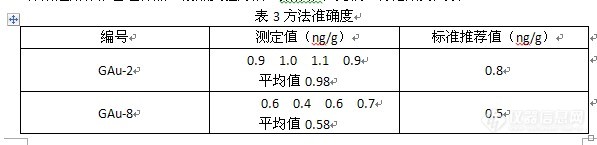
痕量金标准物质湿法分析研究摘要:本文采用王水封闭溶样,活性碳吸附柱原理,将泡沫塑料挤入玻璃吸附柱颈内,将痕量金标准样品溶液进行动态过滤吸附,吸附后的泡沫塑料直接用硫脲解脱,解脱后的溶液用石墨测定。方法精密度再7.1%,准确度满足地质实验室痕量金标准物质质量要求。关键词:痕量金标准物质,动态吸附,泡沫塑料金的泡沫塑料吸附富集分离已是金的富集分离法中应用最广泛的方法之一。泡沫塑料吸附石墨测定痕量金是分析痕量金中最常用的方法,该方法操作简单,效率高,已广泛应用于地质实验实验室中。在常规的预处理过程中,泡沫塑料直接放于试样溶液中振荡吸附,吸附率在80%左右,对于普通样品的测定,可能不会造成分析数据的超差,但是对于标准物质的测定,就很可能超差,鉴于此,本文在实验的基础上,采用活性炭吸附的原理,用泡沫塑料挤入吸附柱颈内动态吸附,实验证明,该方法具有更高的精密度和准确度。1 实验1.1主要仪器及试剂Z-5000型原子吸收仪动态吸附装置封闭溶样装置(水浴锅、聚四氟乙烯溶样罐,振荡器)盐酸、硝酸,氟化氢氨、抗坏血酸、硫脲均为分析纯金标准溶液(0.1µg/ml,10%王水介质)1.2实验方法称取10.0g样品,于高温炉650℃烧2个小时,将样品到倒入聚四氟乙烯溶样罐中,加入少量氟化氢氨润湿,加入25ml王水,盖上盖,振荡摇匀,放入水浴锅中水浴90分钟。冷至室温,加入70-80ml水。倒入预先装好的吸附柱中动态吸附,吸附后的泡沫塑料,放入预先加有1%的硫脲的试管中,水浴40分钟,挤出泡沫塑料,溶液上石墨炉测定。1.3标准系列配制移取0,0.2、0.5,1,2ml金标准溶液(0.1µg/ml)于10ml试管中,用1%硫脲定容到10ml,这样得到0,2,5,10,20ng/ml的标准系列。配制好的标准,随样品放入水浴锅内水浴(空白除外)。1.4吸附柱的安装 将市售的泡沫塑料切成3×1×1cm,用10%盐酸煮沸10min,洗净,然后放入玻璃吸附柱的颈内,装上布氏漏斗,在漏斗上面放一张滤纸和一层滤纸浆。见图1。 图1 吸附装置http://ng1.17img.cn/bbsfiles/images/2011/07/201107032232_302914_1601823_3.jpg1.5仪器工作条件http://ng1.17img.cn/bbsfiles/images/2011/07/201107032239_302918_1601823_3.jpg2结果与讨论2.1标准系列 配置一份标准系列,随样品一起水浴,采用吸光度法测定,制作曲线,测定结果见图2 图2 标准曲线http://ng1.17img.cn/bbsfiles/images/2011/07/201107032241_302921_1601823_3.jpg2.2普通吸附与动态吸附比较称取标准管理样,分别按照常规直接将泡沫塑料放入样品溶液中振荡吸附,动态吸附后,用石墨炉测定,将结果列入表1中。http://ng1.17img.cn/bbsfiles/images/2011/07/201107032242_302922_1601823_3.jpg从表1可以看出,采用动态吸附柱,由于吸附过程加入了过滤装置,泡沫塑料吸附的是没有残渣的溶液,吸附率明显提高,且与标准推荐值很接近。2.3方法精密度称取标准管理样品GAu-9,12份,按照实验方法,用石墨炉原子吸收测定,计算标准偏差,结果列入表2中。http://ng1.17img.cn/bbsfiles/images/2011/07/201107032246_302924_1601823_3.jpg2.4方法准确度http://ng1.17img.cn/bbsfiles/images/2011/07/201107032247_302926_1601823_3.jpg3结论 采用常规的分析方法富集分离痕量金标准物质,常常出现测定值偏低的情况,采用封闭溶样,泡沫塑料动态吸附柱吸附,石墨炉测定痕量金标准物质,不仅提高了吸附率,而且大大提高了标准物质分析的准确度和精密度。
讨论: 工业乙炔中痕量有机气体、无机气体分析 [url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]法成份O2N2 CO2 CO CH4C2H6C2H4C3H8 丙酮C2H2 检测限ppm250.50.50.51112090%--99.99% 六通阀进样,反吹,氢气作载气,1m5A分子筛,分离O2、N2 ,TCD检测;2m Porapark Q ,N2 作载气,分离CO2、CO、CH4 ,甲烷转化炉,FID检测;十通阀进样,Porapark Q预柱,N2 作载气,中心切割,POLT Al2O3 分离,FID检测。上述只是个人意见,不足之处还望批评指正!!
我最近从是高纯气体制备的研究,想跟大家讨论讨论其中痕量杂质的分析检测方法,我所了解到的ICP能进气体样,测定其中的杂质元素,色谱应该也可以,其他还有什么办法没有
我发现很多气体分析需要直接进样的,买的标气都需要稀释的,那你们的实验室是怎么稀释标准气体的?如果GC分析的时候用得是曲线酪校准定量的,你们又是怎么做这种T度的稀释的呢?
请问应该如何稀释标准气体
买了一瓶标准气体,想用气象色谱和该标准气体做浓度曲线,怎么稀释标气呢?http://simg.instrument.com.cn/bbs/images/default/em09511.gif
[color=#444444]标准气体用100ML玻璃针取10mL,20,40 稀释成3个浓度,请问标准稀释后的浓度怎么计算[/color]
各位我想问一下,在实验室想用气相色谱法做甲烷不同浓度的标准曲线,甲烷标准气体怎么稀释呢?请各位帮帮忙,非常感谢。
做气相时常用到痕量级的标准,比如:要将1000ppm的标准储备溶液,配置成5ppb浓度的,按用传统移液管的话,需要稀释好多次,又麻烦又浪费大量的溶剂。而理论上如果用移液器的话,由于它可以移取很小体积,几乎可以一两步就配成了,又快又节约溶剂。我现在不知道的是,它的精确度达不达得到要求啊,有没有人做过比对、或者用这种配法参加过考核并通过了的吗

一个不成熟的东西,供大家参考摘要本文利用微波消解-塞曼效应-GFAAS法测定了人体头发中痕量As、Cd.方法快速,回收率分别为As82.2%~114.1%、Cd 88.4%~112.2%,RSD分别为5.3%、4.1%。结果与标准物质数据相符。关键词微波;GFAAS;头发;As;Cd;中图分类号:O657.32 文献标识码:A 文章编号: 测定人体内微量元素在临床上有重要的价值。利用微波场中分子极化和离子导电效应使样品消解的高压微波消解技术与原子吸收测定由于其显著的优点在近年来得到了迅速的发展。本文用儿童头发对少见于文献的微波消解-石墨炉方法测定As、Cd元素测定进行了探讨,效果满意。1 试验部分1.1 仪器varian SpectrAA 880型原子吸收分光光度计(美国varian公司)。MSP—100D型微波消解仪(北京雷明公司)。玻璃仪器,使用前用硝酸(3+1)浸泡24小时,用去离子水冲净,晾干,备用。1.2 试剂1.2.1标准溶液 As、Cd标准储备溶液浓度为1mg·ml-1。标准使用溶液、标准工作溶液按要求稀释至规定浓度。1.2.2其它试剂国家标准物质:脱脂奶粉,编号GBW 08509;牛肝,GBWE 080193基改剂1:0.2mg·ml–1钯溶液;[font=宋体

我单位现有布鲁克波长色散X射线荧光 S8 Tiger 功率4千瓦~~本想借助各位达人的帮助,通过自己的努力,把仪器充分利用一下:把土壤及沉积物(初步独立建立了能测准近30种元素的曲线,今后还会根据大家所教和所学进一步建立能测准更多元素的标准曲线),环境空气滤膜(用于雾霾源解析),作物小麦玉米大米等。 今天看到吉昂老师的教材说,痕量分析波长色散仪器更多,顿时失去信心。废话这么多,想请教大家两个问题:1.同价位的XRF,究竟那个对痕量元素监测更有优势,我们现有仪器能实现痕量元素1.0甚至更低如0.2 mg/kg(粮食标准标准限值)以下的检出下限吗?2.我们是基层环境监测部门,根据我单位现有WDXRF,如何实现0.2mg/kg甚至更低检出下限呢?需要配置哪些XRF东西?~~不考虑钱的问题。http://ng1.17img.cn/bbsfiles/images/2017/01/201701191656_648205_1699201_3.jpg
请问大家做海水痕量金属(Cu,Zn,Pb等)含量检测的时候使用的标准品都是从哪里采购的?价格如何?谢谢。
[img]http://www.instrument.com.cn/bbs/images/affix.gif[/img][url=http://www.instrument.com.cn/bbs/download.asp?ID=187479]大气中痕量气体污染物的傅里叶变换红外光谱分析.pdf[/url]
岛津GC-2014C的仪器上,在SPL进样口的进样模式里面有不分流进样模式,不分流进样模式是针对于痕量化合物组分的分析,这个痕量有没有标准呢?多少个ppm才算是痕量呢?
二噁英、多氯联苯和氯丙醇的痕量与超痕量检测技术的研究 ——中国疾控中心营养食品所 吴永宁 李敬光 郑明辉 吴文忠 付武胜 张建清 赵云峰 陈左生 庄志雄 邵 兵二噁英、多氯联苯和氯丙醇是当今食品安全和环境科学领域关注热点,PCDD/Fs和PCBs为持久性有机污染物斯德哥尔摩公约中最重要的3类化合物。我国作为签约国在2004年全国人大批准履行,而在履约能力中首先需要具备的超痕量检测能力即使在发达国家也是少数实验室具备,成为一个国家分析水平的标志,已列入卫生部《食品安全行动计划》能力建设考核指标。本研究将稳定性同位素稀释质谱技术应用到我国食品安全和环境分析领域,针对不同目标化合物分别建立了高分辩磁质谱、四极杆低分辩质谱和离子阱串联质谱的标准化检测技术,特别是采用双同位素稀释同时测定4种氯丙醇的技术。通过对EPA1613/1668、FDA 4084和1/RM /31、AOAC2000.1等国际先进方法在食品(鱼、鱼油、奶粉和猪油)和环境(飞灰、土壤和底泥)样品中开展对比筛选和一系列实验室间协同性验证,提出符合国际规范的技术方案,起草并被颁布为国家和环境行业标准4项,起草待颁布标准5项;发表论著30余篇。先后参加涉及未知溶液、鱼、土壤与底泥、飞灰中PCDD/Fs和PCBs(共平面与指示性)的6次国际比对,均取得优异成绩(在136个实验室中名列前45名),使参加测试的二噁英实验室获得国际承认,成为剑桥同位素实验室鱼和土壤标准参考物的定值实验室。该课题意义重大,总体达到国际先进水平,利用双稳定性同位素进行酱油中单氯取代和双氯取代氯丙醇的同时测定方法属于原创性工作、居国际领先水平。在国内首次开展鱼贝类和土壤中污染的二噁英和多氯联苯同系物类型特征指纹库研究和酱油中氯丙醇的大规模调查,获得了中国总膳食二噁英暴露量,不仅证明所建立的方法实用、可行,也为我国履约摸清家底提供依据。首次以起草国身份参加国际食品法典委员会 (CAC) 酱油氯丙醇标准限量和二恶英减低措施的国际标准起草,全面提高了我国的食品安全科学地位。 获2005年中华医学科技奖二等奖
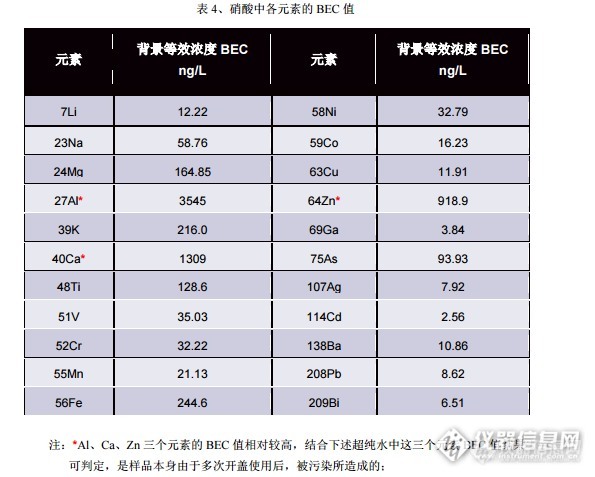
前阵子作为项目,做了高纯硝酸当中的23种杂质元素含量,主要是验证同一方法中运行多种模式以及“碰撞聚焦”的实际效果。 实验结果表明,对于机器背景较高的元素如Na、Mg等以及受Ar基干扰较为严重的元素如K、Ca、Fe等,碰撞聚焦-冷等离子体模式下的“灵敏度/背景”要比单纯地使用“冷等离子体”模式来得更佳。 电感耦合等离子体质谱仪测试高纯硝酸中痕量超痕量元素杂质含量摘要: 采用超纯水稀释高纯硝酸的处理方法,直接测试了高纯硝酸当中的锂、铍、钠、镁、铝、钾、钙、钛、钒、铬、锰、铁、钴、镍、铜、锌、镓、砷、银、镉、钡、铅、铋等 23种元素的含量。实验结果表明,各元素 BEC 值在 0.2~40ng/L 之间,加标回收率在 80~120%,长期稳定性5%。 作为工业和实验室分析当中最为常用的硝酸,其重要作用毋庸置疑。对于光伏、半导体工业来说,这是常见的清洗、反应用酸。其中所含杂质的含量对于产品有着十分重要的影响——例如金属元素过高会导致器件被击穿、P\B 含量则决定着光伏电池的 P/N 型。另外,在ICP-MS 分析当中,由于仪器高灵敏度、样品中目标元素一般为痕量超痕量级别,故样品前处理中最常使用的硝酸也需要做一定程度的杂质管控。 本文参照光伏对于硝酸的标准——SEMI PV16-0611,以标准加入法、“一次进样运行四种模式”测试了高纯硝酸当中的 23 种元素。实验结果表明,对于硝酸中各杂质的背景等效浓度 BEC 值在 0.2~40ng/L 之间,加标回收率均在 80%~120%,2 小时的长期稳定性均在 5%以下。1、 材料与方法:1.1 材料与仪器: 质量分数为 68%的高纯硝酸,随带检测报告表明每种杂质元素含量均不超过1μ g/L;实验用水:Millipore-A20 所制得的超纯水,其电阻率≧18.2MΩ ·cm;ICP-MS2000B:带碰撞反应池和 2 路碰撞反应气,可分别通(He+H2)和(He+NH3),也可根据实际需求配置纯氦气或者其他纯碰撞/反应气;1.2 标准溶液的配置: 锂、铍、钠、镁、铝、钾、钙、钛、钒、铬、锰、铁、钴、镍、铜、锌、镓、砷、银、镉、钡、铅、铋混合标准溶液均由对应的 10mg/L 单标配置而成,各标液购自于 Inorganic Ventures 公司;以重量法经一步稀释成 230.97μ g/L;1.3 前处理方法: 由于 ICP-MS 的离子源 ICP 部分是和大气直接接触的,故等离子体中也有大量的N、O、H,因此硝酸的基体除了酸度影响灵敏度之外,其他的和超纯水并无差别,故前处理上以超纯水直接稀释 10~20 倍即可;考虑到本次样品的纯度仅大致为每元素含量 1ppb 且该样品已多次启封使用,含量已较未开封样品高,故以稀释 20 倍处理。1.4 仪器工作参数: 由于使用了 4 种工作模式,各模式的参数罗列如下:http://ng1.17img.cn/bbsfiles/images/2017/01/201701191701_669011_1638867_3.pnghttp://ng1.17img.cn/bbsfiles/images/2017/10/2016081010231588_01_1638867_3.png2、 结果和讨论2.1 分析模式的选择 由于测试的元素总共有 Li、Be、Na、Mg、Al、K、Ca、Ti、V、Cr、Mn、Fe、Co、Ni、Cu、Zn、Ga、As、Ag、Cd、Ba、Pb、Bi 等 23 个元素。在这些元素当中,可大致分为以下四类:a、 受背景干扰较强但本身并无多原子离子干扰的元素:Li、Na、Mg;这类元素在冷等离子体或者冷等离子体-碰撞反应模式中,背景可以被有效地压制,具体情况如 1.4 的表 2;b、 受多原子离子干扰较为强烈且电离能较低的元素:K(ArH)、Ca(Ar)、Cr(ArC)、Mn(ArN)、Fe(ArO);这类元素在冷等离子体条件下,多原子离子的干扰可以被有效地压制,但本身的灵敏度也比较低。另外,在冷等离子体调谐条件下加入碰撞反应气,除了可以有效提高目标元素灵敏度之外,(He+H2)混合气当中的 H2还可以有效地消除多原子离子的干扰。故这些元素十分适合“碰撞聚焦-冷等离子体”模式;c、 受多原子离子干扰强烈且电离有高有低的元素:Al(CNH、CN)、K(ArH)、Ca(Ar)、Ti(SO、SiOH)、Cr(ArC)、Mn(ArN、ArNH)、Fe(ArO、CaO)、Co(ArF、ArOH)、Cu(NaAr)、Zn(SO2、S2)。这类元素既适合上述的“碰撞聚焦-冷等离子体”,也适合“碰撞-反应模式”。在仔细地比较了“灵敏度-背景”之后,Al、Ti、V、Fe、Zn、Cu 元素选用碰撞反应模式来解决多原子离子的干扰。d、 其他元素:由于并无多原子离子的干扰或者干扰影响很小,同时考虑到灵敏度的问题,故 Be、Ga、Ag、Cd、Ba、Pb、Bi 等元素采用常规模式。综合上述原则,各元素采用的分析模式如表 3:http://ng1.17img.cn/bbsfiles/images/2017/10/2016081010242298_01_1638867_3.png2.2 各分析模式切换的时间和分析总时间 在这四种模式当中,低功率运行的状态一般来说,要比高功率更加不耐受基体。因此如果从高功率向低功率转换,稳定时间要更长一些;另外,通入碰撞反应器和没通碰撞反应气又需要一定的稳定时间。综合以上的因素,实际测试过程中将“碰撞聚焦-冷等离子体”模式放在第一位,其他的依次是“冷等离子体”、“碰撞反应”、“常规”。模式之间切换的稳定时间为:45s、30s、30s、30s。 23 个元素的总分析时间大约为 6 分钟。2.3 内标元素的选择、样品处理方法及分析方法: 任何的动态型分析检测设备,都存在信号的漂移,样品基体也会导致这种情况的发生,因此测试过程当中一般都会采用内标加以校正。但是对于高纯酸而言,由于其待测元素的含量都处于超痕量的水平。如果添加内标,那么无论是内标溶液本身还是添加这个操作,都存在引入污染的风险。另外,实际测试过程中,硝酸样品除了酸度之外其他情况和超纯水十分类似。因此,综合以上因素,测试过程中不用任何的内标。 在前处理的选择上,由于无论是硝酸还是盐酸,如果进行赶酸富集的话,对前处理的环境有较高的要求。考虑到 ICP-MS 的高灵敏度并且这两种类型样品基体和超纯水十分类似,故前处理上以“体积比”的方式用超纯水将待测样品稀释20 倍。测试过程当中,以标准加入法分析各元素含量。2.4 背景等效浓度 BEC 值、加标回收率和 2 小时的长期稳定性: 2.4.1 背景等效浓度 BEC 值为 5%的硝酸信号所对应的浓度值,具体如表 4;另外为验证检测能力,还以标准加入法测试了超纯水中的各个元素http://ng1.17img.cn/bbsfiles/images/2017/10/2016081010260305_01_1638867_3.png 超纯水中各元素 BEC 值测试中,标准曲线为超纯水添加 2.0、5.0、10.0、50.0ng/L。 结果如表 5:http://ng1.17img.cn/bbsfiles/images/2017/10/2016081010263344_01_1638867_3.png 扣除上述超纯水中各元素的浓度值,并乘以稀释倍数,得出硝酸中各元素含量元素 背景等效浓度 BECng/L元素 背景等效浓度 BECng/L7Li 12.22 58Ni 32.7923Na 58.76 59Co 16.2324Mg 164.85 63Cu 11.9127Al* 3545 64Zn* 918.939K 216.0 69Ga 3.8440Ca* 1309 75As 93.9348Ti 128.6 107Ag 7.9251V 35.03 114Cd 2.5652Cr 32.22 138Ba 10.8655Mn 21.13 208Pb 8.6256Fe 244.6 209Bi 6.51值如表 6:http://ng1.17img.cn/bbsfiles/images/2017/10/2016081010290113_01_1638867_3.png2.4.2 加标回收率和长期稳定性: 硝酸的加标回收率以 5%(V/V)硝酸中添加 500ng/L 的混合标准溶液进行测试;同时以 2 小时内测试 21 次的方式测试了长期稳定性。结果分别如表 7 和图 1:http://ng1.17img.cn/bbsfiles/images/2016/
硬聚氯乙烯(UPVC)饮水管材和管件中痕量锡的GFAAS测定研究*(摘自中国公共卫生杂志 2002.11 )浙江省疾病预防控制中心(杭州310009) 鲁 丹 宋国良乐清市疾病预防控制中心 卓岳明摘 要:目的 建立测定硬聚氯乙烯(UPVC) 饮水管材和管件中痕量锡的新方法。方法 应用抗坏血酸作基体改进剂,横向塞曼石墨炉[url=https://insevent.instrument.com.cn/t/Wp][color=#3333ff]原子吸收光谱[/color][/url]法直接测定浸泡液中痕量锡。结果 选定了最佳测量条件,提高了方法的灵敏度、精密度。本法的最低检出量为9.3pg,回收率在94.0 %~101.0 %之间。对锡含量为6.0μg/L的浸泡液连续测定11次的相对标准偏差为4.6 %。结论 本法简便、快速、准确,灵敏度和精密度高。适用于硬聚氯乙烯(UPVC) 饮水管材和管件中痕量锡的测定。关键词:横向塞曼; GFAAS;硬聚氯乙烯(UPVC) 饮水管材和管件;痕量锡中图分类号:R123.5 文献标识码:ADetermination of Trace Sn in Unplastized Polyvinyl Chloride Pipes for Drinking Water Supply by GFAAS L U Dan ,SONG Guo2liang , ZUO Yue2ming. Zhejiang Provincial Center f or Disease Prevention and Cont rol ( Huangz hou 310009 ,China)Abstract: Objective To find new methods to detect Sn in unplastized polyvinyl chloride pipes for drinking water sup2ply. Methods L - Ascorbic acid is used as matrix modifier in the direct Zeeman GFAAS to determine trace Sn.Results Themethod detection limit is 9. 3pg. The recovery is 94.0% - 101.0%. The RSD obtained from the data of 11 determinationsat the concentration level of 6.0μg/L is 4.6%. Conclusion This method is simple,quick, accurate and sensitive. It is a very good methods for measuring Sn in unplastized polyvinyl chloride pipes for drinking water supply.Key words :landscape orentation Zeeman ; GFAAS; unplastized polyvinyl chloride pipes for drinking water supply ;trace Sn 氯乙烯生产过程中为防止其降解,必须加入有机锡化物作为稳定剂,而有机锡化物是剧烈的神经毒物,所以用于饮水管道的聚氯乙烯管材和管件均应检测锡的溶出量。目前,测定锡的方法国外有苯基荧光酮分光光度法、氢化物发生- [url=https://insevent.instrument.com.cn/t/Wp][color=#3333ff]原子吸收光谱[/color][/url]法和石墨炉[url=https://insevent.instrument.com.cn/t/Wp][color=#3333ff]原子吸收光谱[/color][/url]法〔1〕;国内主要是分光光度法〔2,3〕。分光光度法操作烦琐、费时,灵敏度低;石墨炉法虽具有灵敏度高及取量样少的优点〔4〕,但由于受锡易与石墨发生反应、锡的硫化物和氧化物易挥发等因素的影响,使测定方法存在着原子化效率低、基体干扰严重和测量再现性差等问题〔5〕。本研究应用抗坏血酸作基体改进剂,采用热解涂层石墨管和横向塞曼效应扣背景,建立了石墨炉[url=https://insevent.instrument.com.cn/t/Wp][color=#3333ff]原子吸收光谱[/color][/url]法直接测定硬聚氯乙烯(UPVC)饮水管材和管件中痕量锡的方法。本法的特征质量为7.3pg,用于实际样品分析取得了满意的结果。1 实验部分1.1 仪器与试剂 美国热电SOLAAR M6型[url=https://insevent.instrument.com.cn/t/Wp][color=#3333ff]原子吸收光谱仪[/color][/url],带GF95型石墨炉,FS95自动进样装置;美国热电热解涂层石墨管及锡空心阴极灯。锡标准储备液:500μg/ml(冶金部钢铁研究总院提供,GSBG62042 - 90)。锡标准工作溶液:5.0μg/ml。锡标准工作母液:临用时用1%的硝酸,将5.0μg/ml锡标准工作溶液稀释成10.0μg/L。基体改进剂:100g/L 抗坏血酸(分析纯)。硝酸(优级纯)。浸泡水:取25ml 0.04mol/L碳酸氢钠缓冲液、25ml 0.04mol/L钙硬度储备液及0.025mol/L氯储备液,用纯水稀释至1L 。1.2 仪器工作条件 波长224.6nm,光谱通带宽0.5nm,灯电流10mA,保持气体氩气流量0.2L/ min(原子化时停气),进样体积20μL,峰高吸光度定量,横向塞曼扣背景,标准曲线法计算。石墨炉升温程序:干燥90℃,20s,斜坡升温(以10℃/s的速率升温),120℃,15s,斜坡升温(以10℃/ s的速率升温);灰化1000℃,20s,斜坡升温(以150 ℃/s 的速率升温);原子化2300℃,3s,快速升温;清除2500℃,3s,快速升温。1.3 试验方法1.3.1 样品处理 用浸泡水充满受试水管,两端用包有聚四氟乙烯薄膜的干净软木塞或橡皮塞塞紧,在25 ℃±5 ℃避光的条件下浸泡24h ±1h。1.3.2 标准工作曲线 仪器自动将10.0μg/L 的锡标准工作母液用0.5%硝酸稀释成1.0,2.0,4.0,6.0,8.0,10.0μg/L的锡标准系列,自动进样前,与10%抗坏血酸10μl混匀,按仪器工作条件测定。1.3.3 样品测定 吸取10ml浸泡液,加入50μl浓硝酸,摇匀,自动进样前,与10%抗坏血酸10μl 混匀,按仪器工作条件测定。2 结果与讨论2.1 基体改进剂的选择 本法分别试验了以抗坏血酸、硝酸钯、硝酸铵、硝酸镁等试剂作基体改进剂,结果见图1。由图1可以看出:硝酸钯和硝酸镁的改进效果不明显,硝酸铵次之,而抗坏血酸的基体改进效果颇佳,它大大提高了测定锡的灵敏度。同时试验表明,抗坏血酸作基体改进剂后基本消除了基体干扰,背景吸光度值由0.5以上降到0.05以下;提高了测量重现性,测定10.0μg/L 锡标准溶液的RSD由23%降为3.3%。这主要是由于抗坏血酸热分解生成的大量气态碳能增强石墨炉内气氛的还原能力,从而导致锡化合物更完全的原子化。故本法选择抗坏血酸为基体改进剂。2.2 石墨管的选择 分别选用热解涂层石墨管和普通石墨管测定锡含量为10.0μg/L浸泡液(10%抗坏血酸作基体改进剂),结果表明,热解涂层石墨管的灵敏度比普通石墨管高2倍,RSD从17%下降到3.3%,石墨管使用寿命从100多次提高到600多次。所以测定锡宜采用热解涂层石墨管。 图1 几种基体改进剂测定UPVC中锡含量的比较2.3 最佳灰化温度的选择(图2) 由图2 可见,用石墨炉[url=https://insevent.instrument.com.cn/t/Wp][color=#3333ff]原子吸收光谱[/color][/url]法测定浸泡液中痕量锡,未加基体改进剂时,灰化温度超过600℃,锡即有较多的损失,且测量的重现性较差;加入基体改进剂后,灰化温度升至1000℃~1300℃时,未见锡有明显损失,灵敏度有较大提高,测量的重现性较好;但灰化温度升至1400℃时锡有明显损失。但因1000℃时灵敏度与1100℃~1300℃时差不多,而吸收峰形比1100℃~1300℃好,拖尾现象最小,故选择1000℃为测定时的灰化温度。 —◆—10.0μg/ L 锡标准溶液(10%抗坏血酸作基体改进剂)—■—10.0μg/ L 锡标准溶液图2 灰化温度与吸光度的关系2.4 最佳原子化温度的选择 用10%抗坏血酸作基体改进剂,10.0μg/L锡标准溶液在1600℃~2500℃范围内进行最佳原子化温度的选择试验,结果见图3。由图3可见,原子化温度2200℃时出现平台,但因2300℃时灵敏度高,吸收峰形最好且拖尾现象最小,故选择2300℃为最佳原子化温度。2.5 背景扣除方式的选择 用锡含量为10.0μg/L的浸泡液(10%抗坏血酸作基体改进剂),试验了氘灯扣背景和横向塞曼扣背景,结果表明,横向塞曼扣背景的灵敏度比氘灯扣背景高1.5倍,RSD 从9.6%降到3.3% ,背景已基本扣除。故测定锡时宜采用横向塞曼扣背景。 图3 原子化温度与吸光度的关系2.6 干扰试验 本试验在10.0μg/L锡标准溶液中分别加入1.0mg/L的铅、镉、银、铜、汞、镍等元素,结果表明对锡的测定均无干扰。2.7 标准曲线的线性关系 标准曲线的回归方程为Y =0.0091X + 0.0185,r = 0.9994,可见锡标准溶液的浓度在0.00~10.0μg/L 范围内线性关系良好。2.8 方法的特征质量和检出限 本法的特征质量为7.3pg。空白连续测定11次,按3倍标准偏差除以标准曲线的斜率,得出方法的最低检出量为9.3pg。2.9 回收率和精密度试验 对锡含量为6.0μg/L 的样品连续测定11次的RSD为4.6%,Abs为0.068。在样品中分别加入2.5,5.0,10.0μg/L锡标准溶液, 回收率在94.0%~101.0%。2.10 本法与微分电位溶出法的比较 分别用本法及微分电位溶出法〔6〕测定了16种不同的UPVC饮水管材和管件中的锡,对测定结果进行统计学处理,经t检验,P 0.05,即两种方法的测定结果差异无显著性。2.11 样品分析 用本法检测日常送检的UPVC管材和管件中的痕量锡54份,结果锡含量均0.002mg/L (合格,国家限量标准为≤0.002mg/L)。
1 范围本方法适用于硅酸盐岩石及水系沉积物、土壤等成分与硅酸盐岩石成分相近的试样中痕量硒的测定。方法检出限为0.01μg/g,测定范围为0.033~10μg/g。2 原理样品与活性炭混匀,在750℃温度下焙烧,用MgO-Na2CO3捕集,水提取后硒进入溶液与绝大多数共存元素分离。在4moL/L盐酸介质中,样品中的六价硒被硼氢化钾(KBH4)还原成四价硒,进而生成气态的硒化氢(SeH4),由载气(氩气)载入石英炉进行原子化,同时被硒的特种空心阴极灯激发出荧光,其荧光强度与硒含量成正比,经测定荧光强度值求出样品中的硒含量。其化学反应式如下:KBH4+3H2O+HCl→H3BO3+KCl+8H°Se+4+8H°→SeH4↑+2H2↑SeH4 Se+2H2↑对测定Se严重干扰的元素有:Cu、Ag、Au、Pt、Pd、As、Sb、Bi、Hg、Sn以及铁盐存在时Te的干扰。这些元素的含量在不大于下述标出值时,经焙烧后,不干扰0.02μg/mLSe的测定。Cu(10mg),Sn(1mg),As、Sb、Bi、Hg(5mg),Te(500μg),Ag、Au(100μg),Pd、Pt(10μg)。实验表明,样品经焙烧富集分离后,一般样品中共存的元素均不干扰硒的测定。3 试剂3.1 盐酸。3.2 盐酸,4moL/L(即1+2)。3.3 硝酸。3.4 高氯酸。3.5 氧化镁。3.6 碳酸钠。3.7 氢氧化钾。3.8 硼氢化钾,8g/L,2g/L KOH介质,现配现用。3.9 甲基橙水溶液:0.5 g/L。3.10 活性炭:光谱纯。3.11 硒标准溶液3.11.1 硒标准储备液称取0.1000g硒(光谱纯金属),置于100mL烧杯中,加10mL硝酸,低温加热溶解后,加3mL高氟酸,蒸到开始冒白烟取下。冷却后用去离子水吹洗杯壁并蒸至刚冒白烟。加5mL4moL/L的盐酸溶解,移入1L容量瓶中,用4moL/L的盐酸稀至刻度,摇匀备用。该储备液浓度为ρ(Se)=100μg/mL。3.11.2 硒标准工作液用4moL/L的盐酸将上述硒标准储备液逐级稀释成ρ(Se)=10μg/mL;ρ(Se)=1μg/mL;ρ(Se)=0.1μg/mL;ρ(Se)=0.02μg/mL的硒标准工作液。3.12 捕集剂按1∶4的质量比例称取一定量的氧化镁和碳酸钠固体试剂,用玛瑙研钵研匀,备用。4 仪器4.1 原子荧光分析仪。4.2 高性能特种硒空心阴极灯。4.3 自动定量加液装置。5 试样制备5.1 试样粒度应小于74mm。5.2 试样应在105℃预干燥2h~4h,置于干燥器中,冷却至室温后称取。5.3 对易吸水的岩石,应取空气干燥试样,在称样的同时按GB/T14506.1进行吸附水的测定,最终以干态计算结果。6 操作步骤6.1 设定仪器工作参数灯电流100mA;负高压280V;载气流量1200mL/min;积分时间9s;加液时间6s;炉温850℃;炉高6mm。6.2 实验方法吸取5mL酸度为4moL/L盐酸的0.02μg/mL硒标准工作液于氢化物发生器内,盖上发生器盖,启动开关。待硼氢化钾加入后,生成的SeH4由氩气载入石英炉进行原子化,同时被硒的特种空心 阴极灯激发出荧光,荧光强度值由数字表头显示。6.3 空白试验试样空白应随试样分析同时按试样测定(6.4)分析步骤进行。6.4 试样测定称取1g试样(精确至0.0001g)样品于刚玉坩埚内与0.15g活性炭混匀,依次覆盖0.3g氧化镁,1.2g捕集剂。放入马弗炉内,从低温逐渐升高至750℃,保持40min,取出冷后,在聚四氟乙烯烧杯中用水提取熔块,洗出坩埚,控制体积为20mL~30mL,在电热板上煮沸20min。冷却后移入25mL比色管中,用水稀释至刻度,摇匀,澄清。分取15mL清液于聚四氟乙烯烧杯中,加1滴甲基橙,用盐酸调至溶液呈红色。在电热板上蒸至体积约5mL,加入3mL盐酸(3.1),继续加热至溶液体积剩约3mL时取下。转入20mL比色管中,用4moL/L盐酸溶液稀释至刻度,摇匀。取出部分溶液按实验方法测定硒的荧光强度值,并计算其含量。6.5 工作曲线6.5.1 进样体积为5mL时,硒在0.001~0.06μg/mL范围内呈线性关系。分别移取0.1μg/mL硒标准工作液(3.11.2):0.00,0.2,1.0,2.0,4.0,8.0,12mL于20mL比色管中,用4moL/L盐酸稀至刻度,摇匀。配成的标准系列溶液中分别含0.000,0.001,0.005,0.01,0.02,0.04,0.06μg/mL的硒。按实验方法(6.2)的操作步骤测得荧光强度值,绘制工作曲线。6.5.2 进样体积约为2mL时,硒在0.01~0.3μg/mL范围内呈线性关系。分别移取1μg/mL硒标准工作液(3.11.2):0.00,0.2,0.6,1.0,2.0,4.0,6.0mL于20mL比色管中,用4moL/L盐酸稀释至刻度,摇匀。配成的标准系列溶液中分别含0.00,0.01,0.03,0.05,0.1,0.2,0.3μg/mL的硒。按实验方法(6.2)的操作步骤测得荧光强度值,绘制工作曲线。6.5.3 空白试验按实验方法(6.2)的操作步骤,同时作试剂的空白试验。7 结果计算按下式(1)计算硒的含量:w(Se)= mG……(1)式(1)中:w(Se)——样品中Se的质量分数,mg/g;m——试样减空白后荧光强度值在工作曲线上查得的Se质量,μg;G——被测定样品的质量,g。8 精密度对样品中Se含量水平为0.036μg/g和0.089μg/g测量11次,求得RSD分别为10%和5.8%。9 参考文献9.1 任萍,张勤,张锦茂. 焙烧富集分离-氢化物原子荧光测定地质物料中痕量硒. 分析试验室. 1994,13(14):65-67.9.2 郭小伟,张文华,杨密云. 氢化物-无色散原子荧光法测定地质样品中微量硒及碲. 岩石矿物及测试. 1983,2(4):288.9.3 张锦茂,范凡,任萍. 氢化物-原子荧光法测定岩石中痕量硒的干扰及消除. 岩矿测试. 1993,12(4):264.9.4 地球化学标准参考样研制组. 地质专报九一,地球化学标准参考样的研制与分析方法GSD1-8. 北京:地质出版社. 1986,225.
在JJG693-2011《可燃气体检测报警器中》5.1.2.1气体标准物质中,写到“若仪器未找到计划所测气体的种类,可采用异丁烷或者丙烷气体标准物质,标准气体的浓度约为满量程的10%、40%、60%及大于报警设定点浓度的气体标准物质”中的“大于报警设定点浓度的气体标准物质”应该怎么理解,我们在下厂检定的过程中,发现一般报警器的报警设定点一般都在20%至30%之间,规程中只说的大于报警设定点浓度的气体标准物质并没有说大于的范围在多少之间,那么40%或60%的气体标准物质就满足了该要求。是否不一定要配标准气体稀释装置,希望版友参与讨论!
做化学分析都知道,经常会说到痕量分析一词,但到底其标准定义是什么? 有谁知道,出处在那里?
实验室用FPD做硫化氢含量,新人求助,操作过程做硫化氢气体标准曲线具体怎么稀释,现在都是用全玻璃注射器稀释,具体操作是什么,最高浓度是多少
请教各位老师,买了一瓶标准气体,想用标气做浓度曲线,怎么稀释呢?
如题,我们用的赛默飞ICAP Q做人发中元素含量,方法是微波消解头发,然后进样。现在的问题是,《一》如果我们采用直接配制标准溶液,不加基体的话,我们的曲线能做的很好,基本上在三个9以上,但是测定标准人发的值,一般都是证书上值得2-3倍,将样品溶液稀释10倍之后结果也差不多。《二》如果我们采用标准溶液中加标准人发做基体匹配做曲线,曲线基本上不成线性。很困惑,不知道有哪位做ICP-MS方面的专家能帮忙解答一下。PS:内标溶液在线添加,10ppb;标液浓度(ppb)0.02,0.05,0.2,0.5; 2,5,20,50,100 。痕量(Pt,Tl,Th,Co等),微量(Al,Se,Fe,B,I等),常量(Na,K,Ca,Cu,Zn等)

痕量元素分析一站式解决方案软件,提供赛默飞的AA,ICP,[url=https://insevent.instrument.com.cn/t/yp][color=#3333ff]ICPMS[/color][/url]的相关资料查询,免费下载试用。软件功能:1 输入故障代码或关键词可查询相关故障解决案2 查询相关标准,当有新检测项目时,可以通过简单的筛选,即查询到相关的检测标准。3. 提供两项协助功能,通过加微信可获得协助信息。







 我要推广仪器
我要推广仪器
 下载APP
下载APP