据8月28日的《科学》杂志报道说,蚕虫驯养已经有1万多年历史了。蚕为人类提供了宝贵的丝绸和蛋白。但是,现在对蚕基因进行序列测试还为人们提供了一张有关这些随时会为我们提供如此多宝贵物质的昆虫的基因变异图。由西南大学、深圳华大基因带领的国际研究团队为29种家蚕和11种野蚕世系的基因组成功地进行了测序并找到了这些世系之间的差别。共获得了40个家蚕突变品系和中国野桑蚕的全基因组序列,共测632.5亿对碱基序列,覆盖了99.8%的基因组区域,是多细胞真核生物大规模重测序研究的首次报道;绘制完成了世界上第一张基因组水平上的蚕类单碱基遗传变异图谱,这是世界上首次报道的昆虫基因组变异图。科学家还发现了驯化对家蚕生物学影响的基因组印记,从全基因组水平上揭示了家蚕的起源进化。 研究发现,家蚕很明显地在基因上与其野生对应物不同,但即使在各家蚕世系之间,它们仍然维持着大量的变异性。这提示,家蚕只经历了一次牵涉有大量个体的单一且短暂的驯养过程,并在此后在家蚕与野蚕种群之间很少有基因流动。研究人员还能够识别出特别的能够增进丝的生产、蚕虫的繁殖和生长的基因(这些基因很可能是被人类挑选出的)。他们甚至还寻找到了在驯养过程中由蚕虫所获取的行为特征,例如极端的拥挤和容忍人的靠近和操作,以及它们在驯养过程中所丧失的如逃逸及躲避掠食者和疾病等的特征。(
无需进行文库制备,所用DNA样本比标准方法更少2012年12月13日 来源: 中国科技网 作者: 陈丹 中国科技网讯 据物理学家组织网12月12日(北京时间)报道,英国研究人员简化了基因组测序的标准流程,首次无需进行文库制备便完成了DNA(脱氧核糖核酸)单分子测序,而且新方法只要很少量的DNA就能获得序列数据,用量可低至不到1纳克(10亿分之一克),仅为常规测序方法的500分之一到600分之一。 文库制备是指从测序前基因组样本中提取不同长度的DNA片段,这一过程不仅费力、费时,还会浪费DNA,而新技术能极大地减少DNA的损耗,并缩短测序时间。 该研究论文的第一作者、英国威康信托基金会桑格研究所的保罗·库普兰说:“我们用这种方法对病毒和细菌的基因组测序后发现,即使在相对较低的水平,我们也能够确定所检测的是何种有机物,不论样本中是否存在特定的基因或质粒(这对于确定抗生素耐药性很重要),或者其他信息,如对特定DNA碱基的修改等。”他表示,一旦技术得到优化,将在快速、高效地识别医院和其他医疗场所中的细菌和病毒方面具有很大的应用潜力。 研究小组利用第三代单分子测序系统PacBio RS演示了这种简化的直接测序方法。他们仅仅用800皮克(千分之一纳克)DNA来分析一个生物体的基因组,尽管测序仪只读取了基因组的70个序列片段,相对于常规测序方法获得的数据来说不过是很小的一部分,但这些信息足以让研究人员确定他们所检测的生物体的品种。 这项技术也使得科学家能够对此前无法识别的宏基因组(也称微生物环境基因组)样本中的生物体进行确认。“为微生物测序,首先需要能够在实验室中培养它们。”论文的主要作者、英国巴布拉汉研究所的塔米尔·钱德拉说,“这不仅耗费时间,而且有时候微生物不生长,为它们的基因组测序极其困难。”他表示,新方法可以直接对微生物测序,短时间内便可确定其“身份”。 论文的另一主要作者、威康信托基金会桑格研究所的哈罗德·斯维尔德洛说:“我们的技术可以在对所测序列没有任何先验知识、没有特定微生物试剂的条件下,在很短的时间内操作,这是一种很有前途的替代手段,可应用于控制感染等临床需要。”(记者陈丹) 总编辑圈点 长久以来,基因测序等围绕基因科学所展开的研究,都被人们贴上了从本源上解开人体生命奥秘、彻底解除遗传疾病威胁等殷切的标签。多国为提高社会健康水平,都开展了解码国民DNA的活动,有些甚至覆盖全基因组。然而,面对由30亿个碱基对构成的人类基因组,精确测序注定将是一场浩大而又漫长的工程。如何能快速、准确地将海量DNA数据转化为有帮助的实用信息,已经成为该领域科学家们面临的重大挑战之一。因而我们说,英国科学家此番取得的突破,不管是从整个学科研究的方法论层面,还是从临床应用的角度,都提高了基因研究服务于人类的速度。 《科技日报》(2012-12-13 一版)
基因组测序和序列的组装,为快速研究该致病菌株的致病机理创造了条件。与此同时华大基因与德国汉堡-Eppendorf医疗中心合作,也宣布完成了对致病菌株的测序工作。Guenther说:"在有限的时间里完成了对微生物的全基因组测序,极大的方便了研究者从一个整体的水平上去研究微生物,进而揭示在这些目标微生物的基因组究竟发生了哪些改变。"事实上也的确如此,科学家根据从基因组测序的数据所获得的证据,将本次的致病型大肠杆菌鉴定为致病型大肠杆菌的一个新杂交品种,并且携带了一些抗性基因。"从宏观的基因组水平上来研究这类细菌,将在很大程度上革新我们对传染病暴发的认识,3-4天内完成对某种微生物的全基因组测序及基因标注,将会开启一个新的研究领域。"在新奥尔良召开的美国微生物学会年度会议上,一些研究者指出,分子鉴定的方法正被用来打造基因组传染病学这一领域,基因组传染病学致力于重构传染病暴发的过程,以求在将来能够对传染病能进行实时有效的监控和快速反应。
科学家首次测序癌症患者基因组美国科学家近日首次成功测序了一个癌症患者的基因组,这一开创性工作为利用新方法揭开癌症的遗传学基础创造了条件。相关论文发表在11月6日的《自然》(Nature)杂志上。测序的基因组来自于一位女性,50多岁死于急性骨髓性白血病(AML)。美国华盛顿大学的研究人员利用来自皮肤样本的遗传材料,测序了她2套染色体的DNA,同时根据骨髓样本检测了其肿瘤细胞中的遗传突变。所有样本均采自患者接受癌症治疗前,以防DNA受到进一步损伤。随后,研究人员将患者的肿瘤基因组与其正常基因组进行了比较,以期发现遗传差异。在患者肿瘤基因组中接近270万个单核苷变异中,将近98%同样也在患者皮肤样本的DNA中检测到,这就大大缩小了进一步筛选的范围。研究人员最终在患者的肿瘤DNA中仅发现了10个可能与AML有关的遗传突变,其中8个很罕见,它们所处基因之前从未被认为与AML有关。研究人员还显示,肿瘤样本中的每个细胞拥有9个突变,而且较少发生的那个突变可能是最后形成的。研究人员怀疑,所有这些突变对于患者的癌症都很重要。美国国立人类基因组研究所前任主管Francis Collins说:“首次确定人类癌症基因组的完全DNA序列,并与同一个体的正常组织相比较,这在癌症研究中是一个真正的里程碑。”美国俄勒冈健康与科学大学癌症研究所的Brian Druker说:“虽然这一研究尚不能告诉我们怎样治疗癌症患者,但它是这条路上关键的第一步。它为大规模癌症基因组测序和揭示癌症秘密打下了基础。”目前,研究小组正在测序其他AML患者的基因组,同时他们还计划将这种全基因组方法扩展到乳腺癌和肺癌。
最近,来自美国加利福尼亚大学圣地亚哥分校、克雷格·文特尔研究院和Illumina公司的科学家对现代基因测序算法进行了改良,只需从一个细菌细胞中提取的DNA(脱氧核糖核酸)就可组装成接近完整的基因组,准确率达到90%,而传统的测序方法至少需要10亿个相同的细胞才能完成。这一突破为那些无法培养的细菌提供了测序方法。研究发表在9月18日的《自然·生物技术》网络版上。 实验室无法培养的细菌范围极广,约占99.9%,从产生抗体和生物燃料的微生物,到人体内的寄生菌。它们的生存条件特殊,比如必须和其他菌种共生,或只能生存在动物皮肤上,因此很难进行人工培养。 论文合著者、文特尔研究院的罗杰·拉斯肯教授10年前曾开发出一种多重置换扩增(MDA)技术,可对实验室无法培养的细菌测序,能恢复70%的基因。其工作原理是对一个细胞的基因片断多次复制,直到其数量相当于10亿个细胞那么多。不过,这种技术却给测序软件带来很多麻烦,它在复制DNA时会出现各种错误,而且并非完全统一放大,有些基因组被复制数千次,有一些却只被复制一两次。但测序算法不能处理这些不一致,而是倾向于舍弃那些只复制了少数次的基因,即使它们对整个基因组来说很关键。 加州大学圣地亚哥分校雅各布工程学院计算机科学教授、现代基因测序技术算法创建人帕维尔·帕夫纳和同事改进了这一方法,保留了那些少量复制的基因片断,并用新方法对一个大肠杆菌测序以检验其精确性,发现它能恢复91%的基因,接近传统的培养细胞水平。这已足够解答许多重要的生物学问题,比如该细菌能产生什么抗体。 人体细菌占体重的约10%,它们有些会造成传染病,但也有的能帮助消化,最近研究还发现,它们能改变人的行为方式,比如引诱人吃更多的东西。新方法也有助于科学家理解细菌行为,研究人体内细菌能产生哪种蛋白质和多肽,这些蛋白质和多肽是细菌之间、细菌和宿主之间互相沟通的工具。 研究小组还用新方法对一种以前未曾测序过的海洋细菌进行了测序,获得了相当完整而且能解释的基因组,掌握了它是如何生存和运动的,该基因组将被存入美国国家卫生研究院的基因银行(GenBank)。研究人员表示还将对更多迄今未知的细菌进行测序。
第三张“基因变异图谱”与第二代基因组测序技术——评“千人基因组计划”首期研究成果的医学意义世界上任意两个人的基因99%都是相同的,而恰是那1%不同,负责着个体间的表型差异。《自然》杂志近期披露,当人体内携带有250到300基因变异位点的时候,相关基因就就会“沉默”。甚至,一个人只携带了 50到100基因变异位点,就可能患上某种疾病。10年前,“人类基因组计划”这一耗资30亿美元、历时10余年的伟大科学工程完成之际,人们以为得到了揭开自身生命奥秘的天书,生命科学也划时代地进入了“后基因组时代”。如今看来,当时得到的仅仅是人类基因组的“参考图谱”,对于人群里个体间的基因差异,或是更具医学意义的“基因变异图谱”来说,人们知之甚少。第三张“基因变异图谱”为了探寻个体间的基因差异,科学界在2002年启动了HapMap(人类基因组单体型图谱)计划。Hapmap在2005年完成的“第一张基因变异图谱”含有一百万个“单核苷酸多态性”(SNPs)位点;HapMap在2008年完成的“第二张基因变异图谱”含有三百一十万个SNPs位点。而此次“千人基因组”所公布的一期结果——“第三张基因变异图谱”,已经包含了一千五百万个SNPs位点。今年10月28日,《自然》杂志为此刊出的文章题目为“基于群体规模的基因变异图谱”,鲜明的指出,“千人基因组计划”首期研究成果,其最大优势在于:“第三张基因变异图谱”所采用的样本,针对了“大规模人群”。 远超过此前两张“基因变异图谱”所测定的样本数。绘制“第三张基因变异图谱”的所有数据,是基于两个核心家庭,6个个体的精确基因组测序,179个个体的低覆盖率基因组测序,以及七百多人的蛋白编码区的基因测序。检测人群数目庞大,人种涉及中国人、日本人、西欧人等。因此,第三张“人类基因变异图谱”的问世,可以从更深的层次上了解,种族之间、个体之间的基因差异。更具医学意义的是,对于人群中发生频率在1%以上的基因变异,本次研究的覆盖率达到95%以上。这就意味着:此前Hapmap计划所绘制的两张“基因变异图谱”中,没能涉及的“罕见病”致病基因,可能在“第三张基因变异图谱”中已经被标出。“基因变异图谱”的医学应用随着,“人类基因变异图谱”绘制的日臻完善,和商业化全基因组SNP 分型芯片成本的不断降低,以及新的统计方法和软件的出现, “全基因组关联分析”( Genome-Wide Associat ion Study , GWAS) 越来越多的应用于复杂疾病“易感基因”的确定。今年6月6日,安徽医科大学的张学军教授领衔的团队,通过对中国汉族和维吾尔族人群近2万份样本进行分析,在人类基因组的3个区域内发现与白癜风发病密切相关的4个易感基因。今年8月2日,中***事医学院贺福初院士领衔的蛋白质组学国家重点实验室,通过对大陆5个肝癌高发区的4500多名肝癌病例和对照的研究,发现了肝癌易感基因新区域(1p36.22)今年8月23日,新乡医学院的王立东教授联合国内18家医院,建立了数十万份的食管癌标本资料库,并首次在人类第10号和20号染色体上,发现两个食管癌易感基因(PLCE1和C20orf54)。基因变异有着很强的人种差异,相比国外此领域的研究成果,以上研究成果的临床意义,在于其是针对我国的特有人群。也就是说,以上研究成果在我国的临床上更具医学价值。更为可喜的是,以上研究成果均发表在此领域最为权威的《自然 遗传学》杂志上。我国在利用GWAS需找复杂疾病易感基因领域的研究,已经得到了世界的公认。
来自英国Sanger研究院,Illumina Cambridge公司等处的研究人员发表了题为“Genome Sequencing and Analysis of the Tasmanian Devil and Its Transmissible Cancer”的文章,完成了一种传染性癌症的基因组测序,并从中发现了一些突变,解析了这种癌症的来源,以及如何变得具有传染性的。相关成果公布在Cell杂志上。这种癌症主要发生在世界上最大的肉食性有袋动物:袋獾身上,这种动物也被称为塔斯马尼亚恶魔(Tasmanian Devil),现今只分布于澳大利亚的塔斯马尼亚州。袋獾是袋獾属中唯一未灭绝的成员,其在研究领域最著名的就是袋獾面部肿瘤疾病。袋獾面部肿瘤是一种独特癌症,常出现于袋獾面部或嘴部,但通常会扩散至袋獾的内脏,它与另外一种在犬类中传播的恶性肿瘤是世界上仅有的两种可通过上述方式传播的癌症。这项研究离心机揭示了这种能通过撕咬在动物间传播的肿瘤的奥秘,首次针对一个雌性袋獾的单细胞进行分析。这个雌性袋獾被称为“永恒恶魔(The Immortal Devil)”,因为其死于15年前,但它的DNA仍然在传染癌细胞系中流传。文章的第一作者,Sanger研究院Elizabeth Murchison博士表示,“袋獾癌症是目前发现的唯一一种威胁到整个物种灭绝的癌症”,“通过其测序,将有助于我们整理引发整个袋獾群体癌症的突变。”研究人员从中找到了肿瘤细胞之间的遗传差异,这表明这种癌症在袋獾群体中传播的时候,发生了遗传突变。他们在塔斯马尼亚州不同地区找到了69种不同袋獾的肿瘤样品,构建袋獾面部肿瘤传播的图谱,研究结果表明一些癌症亚型比其它亚型更具有侵染性。Illumina Cambridge公司David Bentley说,“我们发现这种癌症的基因组具有大约两万个突变,这比某些人类癌症中发生的突变更少,这说明癌症变得具有传播性,基因组极度不稳定并不是必要条件”,“追踪这种癌症的进化历史,以及其传播过程,将有助于我们了解这种疾病发生的原因,以及预测其未来的发展。”癌症在个体之间的传播正常来说,会受到免疫系统牛血清蛋白的干涉,因为免疫系统可以鉴别外来组织,这一研究组发现了一些有趣的线索——这种癌症如何能“智斗”免疫系统,比如免疫系统中的一组基因突变。但是还需要更进一步的研究,揭示这种癌症是如何从免疫系统中逃脱出来的。“这项研究十分重要,因为这将会帮助我们理解疾病传播的模式,也有助于疫情的研究,但是我们还需要利用这一基因组测序,更进一步分析这种癌症如何变得具有传染性。癌症具有群体传播性,显示是非常罕见的,我们通过袋獾这一例子来分析这一过程,以防未来在人类身上发生”,Sanger研究院,文章通讯作者Mike Stratton教授说。研究组下一步将进行更多袋獾基因组测序,绘制上千袋獾肿瘤样品基因组图谱,从而更好的了解这种癌症的遗传多样性,并分析癌症与袋獾群体之间的遗传关联性。去年这一研究组在Science杂志上发表文章,发现培养基袋獾面部肿瘤起源于雪旺细胞。他们从分布在澳大利亚塔斯马尼亚岛14处的袋獾群落中采集了25个袋獾面部肿瘤样本,进行基因分析,结果发现,袋獾面部肿瘤起源于雪旺细胞,在大约20年前,袋獾雪旺细胞内的某种基因变异导致了这一癌变。
[align=center][b][font=宋体]利用[/font][font='Times New Roman']MGI[/font][font=宋体]平台对大豆进行全基因组重测序分析[/font][/b][/align][b][font=宋体]摘要[/font][/b][font=宋体][font=宋体]:本研究建立了[/font][font=Times New Roman]MGI[/font][font=宋体]平台全基因重测序的方法。[/font][font=Times New Roman]MGI[/font][font=宋体]平台对大豆的全基因进行重测序结果显示,测序数据质量良好,且与参考基因组比对率较高,符合后续分析要求,对其进行[/font][font=Times New Roman]SNP[/font][font=宋体]和[/font][font=Times New Roman]Indel[/font][font=宋体]的变异检测和注释,此结果说明今后可利用[/font][font=Times New Roman]MGI[/font][font=宋体]平台对其它样品进行全基因重测序分析。[/font][/font][b][font=宋体]关键词[/font][/b][font=宋体][font=宋体]:[/font][font=Times New Roman]MGI[/font][font=宋体]平台;全基因重测序[/font][/font][align=center][font='Times New Roman']Whole genome resequencing analysis of soybeans using the MGI platform[/font][/align][b][font='Times New Roman']Abstract:[/font][font=宋体] [/font][/b][font=宋体][font=Times New Roman]In this study, a method for whole gene resequencing on the MGI platform was established. The results of resequencing the whole genes of soybean by MGI platform showed that the sequencing data was of good quality and had a high comparison rate with the reference genome, which met the requirements of subsequent analysis, and the variation detection and annotation of SNP and Indel were carried out, which indicated that the MGI platform could be used to perform whole gene resequencing analysis on other samples in the future.[/font][/font][b][font='Times New Roman']Keywords:[/font][font=宋体] [/font][/b][font=宋体][font=Times New Roman]MGI platform Whole gene resequencing[/font][/font][font='Times New Roman'] [/font][b][font='Times New Roman']1 [font=宋体]研究背景[/font][/font][/b][font='Times New Roman'][font=宋体]大豆是重要的粮食作物和油料作物,也是人类最主要的植物蛋白来源[/font][/font][font=宋体][font=Times New Roman][1][/font][/font][font=宋体][font=宋体]。我国是野生大豆的发源地,有着极其丰富的大豆种质资源基础,但是育种和产量较其他大豆主产国显得略有不足,究其原因是我国对大豆的研究和发掘力度存在不足,因此,对大豆育成品种的改良势在必行。自[/font][font=Times New Roman]2010[/font][font=宋体]年起,大豆群体水平的重测序也全面开展,在大豆的全基因组变异图谱上也得到了一定的研究进展[/font][/font][font=宋体][font=Times New Roman][2][/font][/font][font=宋体][font=宋体]。本研究利用[/font][font=Times New Roman]MGI[/font][font=宋体]平台对大豆全基因组进行重测序分析,挖掘全基因组水平上的突变。[/font][/font][b][font=宋体][font=Times New Roman]2 [/font][font=宋体]实验仪器[/font][/font][/b][font=宋体]主要实验仪器:[/font][font=宋体][font=Times New Roman]MGISP-960[/font][font=宋体]、[/font][font=Times New Roman]MGIDL-T7[/font][font=宋体]、[/font][font=Times New Roman]DNBSEQ-T7[/font][/font][b][font=宋体][font=Times New Roman]3 [/font][font=宋体]实验结果[/font][/font][font=宋体][font=Times New Roman]3.1 [/font][font=宋体]测序数据质量[/font][/font][/b][font=宋体][font=宋体]根据[/font][font=Times New Roman]MGI[/font][font=宋体]平台的测序特点,使用双端测序的数据,要求[/font][font=Times New Roman]Q30[/font][font=宋体]平均比例在[/font][font=Times New Roman]85%[/font][font=宋体]以上,可以看出大豆重测序数据[/font][font=Times New Roman]Q30[/font][font=宋体]平均比例在[/font][font=Times New Roman]94.72%[/font][font=宋体]以上,说明大豆测序数据质量良好,满足分析要求。[/font][/font][font='Times New Roman'] [/font][font='Times New Roman'] [/font][b][font=黑体][font=黑体]表[/font][font=Times New Roman]1 [/font][font=黑体]测序数据统计表[/font][/font][/b][table][tr][td][align=center][font='Times New Roman']Samples[/font][/align][/td][td][align=center][font='Times New Roman']ID[/font][/align][/td][td][align=center][font='Times New Roman']Clean reads[/font][/align][/td][td][align=center][font='Times New Roman']Clean bases[/font][/align][/td][td][align=center][font='Times New Roman']GC Content[/font][/align][/td][td][align=center][font='Times New Roman']%[/font][font=等线]≥[/font][font='Times New Roman']Q20[/font][/align][/td][td][align=center][font='Times New Roman']%[/font][font=等线]≥[/font][font='Times New Roman']Q30[/font][/align][/td][/tr][tr][td][align=center][font='Times New Roman']P117[/font][/align][/td][td][align=center][font='Times New Roman']P117[/font][/align][/td][td][align=center][font='Times New Roman']169494922[/font][/align][/td][td][align=center][font='Times New Roman']25424238300[/font][/align][/td][td][align=center][font='Times New Roman']36.18%[/font][/align][/td][td][align=center][font='Times New Roman']98.49%[/font][/align][/td][td][align=center][font='Times New Roman']95.27%[/font][/align][/td][/tr][tr][td][align=center][font='Times New Roman']P118[/font][/align][/td][td][align=center][font='Times New Roman']P118[/font][/align][/td][td][align=center][font='Times New Roman']166483906[/font][/align][/td][td][align=center][font='Times New Roman']24972585900[/font][/align][/td][td][align=center][font='Times New Roman']36.47%[/font][/align][/td][td][align=center][font='Times New Roman']98.61%[/font][/align][/td][td][align=center][font='Times New Roman']95.70%[/font][/align][/td][/tr][tr][td][align=center][font='Times New Roman']P119[/font][/align][/td][td][align=center][font='Times New Roman']P119[/font][/align][/td][td][align=center][font='Times New Roman']186127112[/font][/align][/td][td][align=center][font='Times New Roman']27919066800[/font][/align][/td][td][align=center][font='Times New Roman']35.89%[/font][/align][/td][td][align=center][font='Times New Roman']98.57%[/font][/align][/td][td][align=center][font='Times New Roman']95.61%[/font][/align][/td][/tr][tr][td][align=center][font='Times New Roman']P120[/font][/align][/td][td][align=center][font='Times New Roman']P120[/font][/align][/td][td][align=center][font='Times New Roman']192397276[/font][/align][/td][td][align=center][font='Times New Roman']28859591400[/font][/align][/td][td][align=center][font='Times New Roman']36.46%[/font][/align][/td][td][align=center][font='Times New Roman']98.22%[/font][/align][/td][td][align=center][font='Times New Roman']94.72%[/font][/align][/td][/tr][tr][td][align=center][font='Times New Roman']P198[/font][/align][/td][td][align=center][font='Times New Roman']P198[/font][/align][/td][td][align=center][font='Times New Roman']141636468[/font][/align][/td][td][align=center][font='Times New Roman']21245470200[/font][/align][/td][td][align=center][font='Times New Roman']37.11%[/font][/align][/td][td][align=center][font='Times New Roman']98.67%[/font][/align][/td][td][align=center][font='Times New Roman']95.84%[/font][/align][/td][/tr][tr][td][align=center][font='Times New Roman']P199[/font][/align][/td][td][align=center][font='Times New Roman']P199[/font][/align][/td][td][align=center][font='Times New Roman']169468714[/font][/align][/td][td][align=center][font='Times New Roman']25420307100[/font][/align][/td][td][align=center][font='Times New Roman']36.55%[/font][/align][/td][td][align=center][font='Times New Roman']98.60%[/font][/align][/td][td][align=center][font='Times New Roman']95.66%[/font][/align][/td][/tr][tr][td][align=center][font='Times New Roman']P200[/font][/align][/td][td][align=center][font='Times New Roman']P200[/font][/align][/td][td][align=center][font='Times New Roman']155078286[/font][/align][/td][td][align=center][font='Times New Roman']23261742900[/font][/align][/td][td][align=center][font='Times New Roman']37.90%[/font][/align][/td][td][align=center][font='Times New Roman']98.77%[/font][/align][/td][td][align=center][font='Times New Roman']96.14%[/font][/align][/td][/tr][/table][font=Calibri] [/font][font=宋体][font=宋体]样品原始数据碱基质量值可由图[/font][font=Times New Roman]1[/font][font=宋体]看出不存在异常碱基,[/font][font=Times New Roman]6[/font][font=宋体]个大豆碱基测序错误率分布均如图[/font][font=Times New Roman]1[/font][font=宋体]。[/font][/font][align=center][img=,321,]file:///C:/Users/xuxu/AppData/Local/Temp/ksohtml9716/wps1.jpg[/img][font=Calibri] [/font][/align][align=center][b][font=黑体][font=黑体]图[/font] [font=Times New Roman]1 [/font][font=黑体]碱基测序错误率分布图[/font][/font][/b][/align][font=宋体][font=宋体]碱基类型分布检查可用于检测有无[/font][font=Times New Roman]AT[/font][font=宋体]、[/font][font=Times New Roman]GC[/font][font=宋体]分离现象,若有碱基分离现象可能是测序或建库所带来的,并会影响后续分析。高通量所测序为基因组随即打断后的[/font][font=Times New Roman]DNA[/font][font=宋体]片段,由于位点在基因组上的分布是近似均匀的,同时,[/font][font=Times New Roman]G/C[/font][font=宋体]、[/font][font=Times New Roman]A/T[/font][font=宋体]含量也是近似均匀的。因此,根据大数定理,在每个测序循环上,[/font][font=Times New Roman]GC[/font][font=宋体]、[/font][font=Times New Roman]AT[/font][font=宋体]含量应当分别相等,且等于基因组的[/font][font=Times New Roman]GC[/font][font=宋体]、[/font][font=Times New Roman]AT[/font][font=宋体]含量。同样因为重叠等的关系会导致样品前几个碱基[/font][font=Times New Roman]AT[/font][font=宋体]、[/font][font=Times New Roman]GC[/font][font=宋体]不等波动较大,高于其他测序区段,而其它区段的[/font][font=Times New Roman]GC[/font][font=宋体]、[/font][font=Times New Roman]AT[/font][font=宋体]含量相等,且分布均匀无分离现象,如图[/font][font=Times New Roman]2[/font][font=宋体]所示。[/font][/font][align=center][img=,321,]file:///C:/Users/xuxu/AppData/Local/Temp/ksohtml9716/wps2.jpg[/img][font=Calibri] [/font][/align][b][font=黑体][font=黑体]图[/font][font=Times New Roman]2 ATGC[/font][font=黑体]含量分布图[/font][/font][font=宋体][font=Times New Roman]3.2 [/font][font=宋体]与参考基因组的序列比对[/font][/font][font='Times New Roman']3.2.1 [font=宋体]比对结果[/font][/font][/b][font=宋体][font=宋体]将测序得到的大豆样品与参考基因进行序列比对,[/font][font=Times New Roman]bwa[/font][font=宋体]软件主要用于二代高通量测序得到的短序列与参考基因组进行比对,比对结果见表[/font][font=Times New Roman]2[/font][font=宋体],根据比对结果可评估测序数据是否满足后续分析。[/font][/font][align=center][b][font=黑体][font=黑体]表[/font][font=Times New Roman]2 [/font][font=黑体]比对效率统计表[/font][/font][/b][/align][table][tr][td][align=center][font='Times New Roman']Sample_ID[/font][/align][/td][td][align=center][font='Times New Roman']Mapped(%)[/font][/align][/td][td][align=center][font='Times New Roman']Properly_mapped(%)[/font][/align][/td][td][align=center][font='Times New Roman']Averge_depth[/font][/align][/td][/tr][tr][td][align=center][font='Times New Roman']P117[/font][/align][/td][td][align=center][font='Times New Roman']99.99%[/font][/align][/td][td][align=center][font='Times New Roman']98.53%[/font][/align][/td][td][align=center][font='Times New Roman']25.44[/font][/align][/td][/tr][tr][td][align=center][font='Times New Roman']P118[/font][/align][/td][td][align=center][font='Times New Roman']99.99%[/font][/align][/td][td][align=center][font='Times New Roman']98.55%[/font][/align][/td][td][align=center][font='Times New Roman']24.9[/font][/align][/td][/tr][tr][td][align=center][font='Times New Roman']P119[/font][/align][/td][td][align=center][font='Times New Roman']99.99%[/font][/align][/td][td][align=center][font='Times New Roman']98.63%[/font][/align][/td][td][align=center][font='Times New Roman']27.75[/font][/align][/td][/tr][tr][td][align=center][font='Times New Roman']P120[/font][/align][/td][td][align=center][font='Times New Roman']99.98%[/font][/align][/td][td][align=center][font='Times New Roman']98.28%[/font][/align][/td][td][align=center][font='Times New Roman']28.58[/font][/align][/td][/tr][tr][td][align=center][font='Times New Roman']P198[/font][/align][/td][td][align=center][font='Times New Roman']99.99%[/font][/align][/td][td][align=center][font='Times New Roman']98.58%[/font][/align][/td][td][align=center][font='Times New Roman']21.26[/font][/align][/td][/tr][tr][td][align=center][font='Times New Roman']P199[/font][/align][/td][td][align=center][font='Times New Roman']99.98%[/font][/align][/td][td][align=center][font='Times New Roman']98.50%[/font][/align][/td][td][align=center][font='Times New Roman']25[/font][/align][/td][/tr][tr][td][align=center][font='Times New Roman']P200[/font][/align][/td][td][align=center][font='Times New Roman']99.99%[/font][/align][/td][td][align=center][font='Times New Roman']98.13%[/font][/align][/td][td][align=center][font='Times New Roman']23.13[/font][/align][/td][/tr][/table][font=宋体][font=宋体]将比对到不同染色体的[/font][font=Times New Roman]Reads[/font][font=宋体]进行位置分布统计,绘制[/font][font=Times New Roman]Mapped Reads[/font][font=宋体]在参考基因组上的覆盖深度分布图,见图[/font][font=Times New Roman]3[/font][font=宋体]。[/font][/font][align=center][img=,321,]file:///C:/Users/xuxu/AppData/Local/Temp/ksohtml9716/wps3.jpg[/img][font=Calibri] [/font][/align][align=center][b][font=黑体][font=黑体]图[/font][font=Times New Roman]3 Mapped Reads[/font][font=黑体]在参考基因组上的位置及覆盖深度分布图[/font][/font][/b][/align][font=宋体][font=宋体]统计[/font][font=Times New Roman]Mapped Reads[/font][font=宋体]在指定的参考基因组不同区域的数目,绘制基因组不同区域样品[/font][font=Times New Roman]Mapped Reads[/font][font=宋体]的分布图,见图[/font][font=Times New Roman]4[/font][/font][align=center][img=,321,]file:///C:/Users/xuxu/AppData/Local/Temp/ksohtml9716/wps4.jpg[/img][font=Calibri] [/font][/align][b][font=黑体][font=黑体]图[/font][font=Times New Roman]4 [/font][font=黑体]基因组不同区域[/font][font=Times New Roman]Reads[/font][font=黑体]分布图[/font][/font][font=宋体][font=Times New Roman]3.2.2 [/font][font=宋体]插入片段长度检验[/font][/font][/b][font=宋体][font=宋体]通过检测双端序列在参考基因组上的起止位置,可以得到样品[/font][font=Times New Roman]DNA[/font][font=宋体]打断后得到的测序片段的实际大小,即插入片段大小([/font][font=Times New Roman]Insert Size[/font][font=宋体]),它是信息分析时的一个重要参数。插入片段大小的分布一般符合正态分布,且只有一个单峰,[/font][font=Times New Roman]Insert Size[/font][font=宋体]分布图可以展示各个样品的插入片段的长度分布情况。各样品的插入片段长度模拟分布图见图[/font][font=Times New Roman]5[/font][font=宋体]。[/font][/font][align=center][img=,321,]file:///C:/Users/xuxu/AppData/Local/Temp/ksohtml9716/wps5.jpg[/img][font=Calibri] [/font][/align][align=center][b][font=黑体][font=黑体]图[/font][font=Times New Roman]5 [/font][font=黑体]插入片段长度模拟图[/font][/font][/b][/align][b][font=宋体][font=Times New Roman]3.2.3[/font][/font][font='Times New Roman'][font=宋体]深度分布统计图[/font][/font][/b][font='Times New Roman']Reads[font=宋体]定位到参考基因组后,可以统计参考基因组上碱基的覆盖情况。参考基因组上被[/font][font=Times New Roman]reads[/font][font=宋体]覆盖到的碱基数占基因组的百分比称为基因组覆盖度;碱基上覆盖的[/font][font=Times New Roman]reads[/font][font=宋体]数为覆盖深度。基因组覆盖度可以反映参考基因组上变异检测的完整性,覆盖到的区域越多,可以检测到的变异位点也越多。[/font][/font][font='Times New Roman'][font=宋体]覆盖度主要受测序深度以及样品与参考基因组亲缘关系远近的影响。基因组的覆盖深度会影响变异检测的准确性,在覆盖深度较高的区域(非重复序列区),变异检测的准确性也越高。[/font][/font][font='Times New Roman'][font=宋体]另外,若基因组上碱基的覆盖深度分布较均匀,也说明测序随机性较好。样品的碱基覆盖深度分布曲线和覆盖度分布曲线见图[/font][/font][font=宋体][font=Times New Roman]6[/font][font=宋体]。[/font][/font][align=center][img=,321,]file:///C:/Users/xuxu/AppData/Local/Temp/ksohtml9716/wps6.jpg[/img][font=Calibri] [/font][/align][align=center][b][font=黑体][font=黑体]图[/font] [font=Times New Roman]6 [/font][font=黑体]深度分布统计图[/font][/font][/b][/align][b][font=宋体][font=Times New Roman]3.3 [/font][font=宋体]变异检测[/font][/font][font=宋体][font=Times New Roman]3.3.1 SNP[/font][font=宋体]检测与注释[/font][/font][/b][font='Times New Roman'][font=宋体]根据变异位点在参考基因组上的位置以及参考基因组上的基因位置信息,可以得到变异位点在基因组发生的区域(基因间区、基因区或[/font]CDS[font=宋体]区等),以及变异产生的影响(同义非同义突变等)。软件可以使用[/font][font=Times New Roman]vcf[/font][font=宋体]格式文件作为输入和输[/font][/font][font=宋体][font=宋体]出,见图[/font][font=Times New Roman]7[/font][font=宋体]和图[/font][font=Times New Roman]8[/font][font=宋体]。[/font][/font][align=center][img=,321,]file:///C:/Users/xuxu/AppData/Local/Temp/ksohtml9716/wps7.jpg[/img][font=Calibri] [/font][/align][align=center][b][font=黑体][font=黑体]图[/font][font=Times New Roman]7 SNP[/font][font=黑体]突变类型分布图[/font][/font][/b][/align][align=center][img=,344,]file:///C:/Users/xuxu/AppData/Local/Temp/ksohtml9716/wps8.jpg[/img][font=Calibri] [/font][/align][b][font=黑体][font=黑体]图[/font][font=Times New Roman]8 SNP[/font][font=黑体]注释分类图[/font][/font][font=宋体][font=Times New Roman]3.3.2 Indel[/font][font=宋体]检测与注释[/font][/font][/b][font=宋体][font=宋体]根据所有样品在[/font][font=Times New Roman]CDS[/font][font=宋体]区和全基因范围的[/font][font=Times New Roman]Indel[/font][font=宋体]长度进行统计,其长度分布如图[/font][font=Times New Roman]9[/font][font=宋体]。[/font][/font][align=center][img=,355,]file:///C:/Users/xuxu/AppData/Local/Temp/ksohtml9716/wps9.jpg[/img][font=Calibri] [/font][/align][align=center][b][font=黑体][font=黑体]图[/font][font=Times New Roman]9 [/font][font=黑体]全基因和编码区[/font][font=Times New Roman]Indel[/font][font=黑体]长度分布图[/font][/font][/b][/align][font='Times New Roman'][font=宋体]根据样品检测得到的[/font]Ind[/font][font=宋体][font=Times New Roman]el[/font][/font][font='Times New Roman'][font=宋体]位点在参考基因组上的位置信息,对比参考基因组的基因、[/font]CDS[font=宋体]位置等信息,可以注释[/font][font=Times New Roman]Indel[/font][font=宋体]位点是否发生在基因间区、基因区或[/font][font=Times New Roman]CDS[/font][font=宋体]区、是否为移码突变等。发生移码突变的[/font][font=Times New Roman]Indel[/font][font=宋体]可能会导致基因功能的改变,具体注释结果见[/font][/font][font=宋体][font=宋体]图[/font][font=Times New Roman]10[/font][font=宋体]。[/font][/font][align=center][img=,344,]file:///C:/Users/xuxu/AppData/Local/Temp/ksohtml9716/wps10.jpg[/img][font=Calibri] [/font][/align][align=center][b][font=黑体][font=黑体]图[/font] [font=Times New Roman]10 Indel [/font][font=黑体]注释分类图[/font][/font][/b][/align][b][font=宋体][font=Times New Roman]4 [/font][font=宋体]结论[/font][/font][/b][font=宋体][font=宋体]本文基于[/font][font=Times New Roman]MGI[/font][font=宋体]对大豆进行重基因测序,实验结果可看出,大豆样品测序产出数据良好,与参考基因组序列比对率较高,符合后续分析,对其进行变异检测可得到[/font][font=Times New Roman]SNP[/font][font=宋体]和[/font][font=Times New Roman]Indel[/font][font=宋体]的结果。其它研究表明[/font][/font][font=宋体][font=Times New Roman]MGISEQ-2000[/font][font=宋体]全基因组重测序表现性能稳定、质量可靠,在实际应用上有明显的优势和应用价值[/font][font=Times New Roman][3][/font][font=宋体]。对[/font][/font][font=宋体][font=宋体]本次实验说明[/font][font=Times New Roman]MGI[/font][font=宋体]平台对样品进行重测序效果良好,后续可对其它植物进行重测序。[/font][/font][font=宋体] [/font][font=宋体] [/font][font=宋体]参考文献:[/font][font=宋体][font=Calibri][1] [/font][/font][font='Times New Roman'][font=宋体]张永芳[/font],[font=宋体]钱肖娜[/font][font=Times New Roman],[/font][font=宋体]王润梅[/font][/font][font=宋体][font=Times New Roman],[/font][font=宋体]等[/font][font=Times New Roman]. [/font][font=宋体]不同大豆材料的抗旱性鉴定及耐旱品种筛选[/font][font=Times New Roman][J].[/font][font=宋体]作物杂志[/font][font=Times New Roman],2019(5): 41-45.[/font][/font][font=宋体][font=Calibri][2] [/font][font=宋体]邬启帆[/font][font=Calibri]. [/font][font=宋体]基于基因组重测序黄淮海大豆育成品种遗传结构及重要家族遗传基础研究[/font][font=Calibri][D]. [/font][font=宋体]南昌[/font][/font][font=宋体][font=宋体]大学[/font][font=Times New Roman], 2023.[/font][/font][font=宋体][font=Calibri][3] [/font][/font][font=宋体][font=宋体]李伟宁[/font][font=Times New Roman],[/font][font=宋体]刘刚[/font][font=Times New Roman],[/font][font=宋体]周荣等[/font][font=Times New Roman]. MGISEQ-2000[/font][font=宋体]、[/font][font=Times New Roman]HiSeq 2000[/font][font=宋体]与[/font][font=Times New Roman]NovaSeq 6000[/font][font=宋体]平台全基因组重测序数据的比较分析[/font][font=Times New Roman][J]. [/font][font=宋体]中国畜牧杂志[/font][font=Times New Roman],2021,57(11):156-162.[/font][/font]
来源:中国科技网-科技日报 作者:王怡 2013年10月31日 原标题:我自主研发基因测序技术将实现产业化 科技日报讯(记者王怡)2013年国际基因组学大会10月29日在青岛举行。在开幕现场,中国科学院北京基因组研究所与吉林紫鑫药业股份有限公司就合作开发第二代高通量测序系统项目签订投资意向协议,这标志着由中科院自主研发的第二代测序仪项目即将进入市场转化和产业转化阶段。 基因测序技术,自人类基因组计划实施以来长期占据着国际生命科学技术研究的制高点,随着第二代基因测序技术的发展日趋成熟和成本急剧降低,该项技术被越来越多的科研和实践领域所应用,形成庞大市场。目前我国市场上所有高通量测序设备和试剂均来源于进口,据估计仅2013年我国在仪器和试剂上的投入就超过20亿元。 “我们的基因组学研究一直处于世界前列,源于我们最早参与人类基因组测序的工程,但是我们使用的设备一直都依靠国外的进口设备,中国科学院作为国立科研机构,我们有义务自主研制开发基因测序仪打破国外垄断。”中科院北京基因组研究所党委书记杨卫平说。 在中科院资助下,历时两年半时间完成第二代测序仪研发项目,于2011年实现原理样机和性能验收,部分性能指标超越同类进口产品。其后中科院北京基因组研究所自主投入完成该项目的工程化和产品化开发,并形成其自主知识产权群,目前已有9个专利获得授权。 “第二代高通量测序仪的产业化发展是我们的第一步,后面我们希望能有更多的进展,比如在试剂、数据库和后台都能实现国产化。”杨卫平说。
近日,深圳华大基因研究院和美国科学家共同发起“共生体基因组计划”。该计划将对海蛤蝓(又称绿叶海蜗牛)及藻类饵料进行基因组测序。有科学家认为,海蛤蝓可能是“生命之树”中动植物界的交叉点。海蛤蝓的细胞能够从藻类获取叶绿素,进行光合作用,从而为其所有生命活动提供足够的能量,包括繁殖。迄今为止,科学家在海蛤蝓基因组里发现了大约十多种藻类基因,这些基因使这种生物在叶绿素合成通道和碳固定循环中具有集光蛋白质和酶类的功能。随着研究的深入,不断有新的藻类基因在海蛤蝓基因组中被发现。海蛤蝓通过自身内被转移的藻类基因合成叶绿素,进行光合作用。这种神奇的共生现象第一次证明了一套完整的生物合成途径可以从一种多细胞生物传递到另一种多细胞生物。华大基因有关专家表示,通过对藻类和海蛤蝓的基因组进行比较,不仅将在宿主细胞中发现一组能够进行持续光合作用的基因,而且能够找到转移的特性,包括转移基因片段的大小、数量;更重要的是了解这种转移的运行机制。这些发现将对基因组的人工调控和基因治疗新技术的开发产生重大现实意义。此外,这两类生物的基因组测序将有利于比较基因组研究、进化规则、发展生物学及分类学的发展。据悉,这次联合研究是华大基因“千种动植物参考基因组计划”的一部分。该计划将在未来两年内建立1000种动植物的参考基因组序列。在“共生体基因组计划”中,华大基因主要负责测序和生物信息分析工作。《科学时报》 (2010-3-23 A1 要闻)
7月20日,我国科学家宣布“兰花基因组计划”正式启动。两岸科学家将联手对被喻为“植物界大熊猫”的兰科植物进行全基因组测序和生物信息分析,同时对10种最具代表性的兰科植物进行基因表达的转录组测序和分析。 国家兰科植物种质资源保护中心刘仲键教授介绍,对兰科植物的科学研究历史悠久,其成果为达尔文进化论提供了强有力的支持。兰花研究为进化生物学乃至整个生命科学的发展贡献巨大,至今仍是研究生命与进化的理想模式,占有特殊地位。同时,兰花也是世界性濒危物种,是国际公约保护物种的重中之重。兜兰与国宝大熊猫同列为一级保护,其余兰花全部被列入二级以上保护。 清华大学黄来强教授称,兰花全基因组及转录组测序分析,将为人类提供用现代生物学的新技术和理念从分子生物学的层面审视达尔文的研究,为进化生物学和进化论注入新鲜血液。在基因组和转录组的研究基础上进一步结合生物信息、分子生物、蛋白质组、代谢组、生化、生物物理等多学科和研究手段的融合,对加深其基因组结构及功能的了解,揭示兰科的进化,对生命科学研究具有普遍的重要意义。“兰花基因组计划”涉及的不仅是植物学,还将为世界上相关研究提供全新的起点和平台,是对全球基因组科学的又一重大贡献。 “兰花基因组计划”项目,由深圳兰科植物保护研究中心(国家兰科植物种质资源保护中心)、清华大学、深圳华大基因研究院、中国科学院植物所、台湾成功大学等单位科学家共同承担。
http://img.dxycdn.com/trademd/upload/userfiles/image/2013/01/B1357710940_small.jpg梅花因其独特的花香,在很多诗词中成为人们吟诵的对象。那么,它的花香到底来自何处呢?我国科学家从基因组水平,揭示了合成梅花花香中重要成分乙酸苯甲酯的BEAT基因家族34个成员,并构建完成了首张梅花全基因组精细图谱。其研究论文在2012年12月27日《自然—通讯》亮点论文在线发表。我国梅花基因组项目首席专家、北京林业大学教授张启翔率领项目组,选取位于梅花起源中心的西藏野生梅花进行基因组测序,从基因组水平,揭示了合成梅花花香中重要成分乙酸苯甲酯的BEAT基因家族34个成员,在梅花基因组中显著扩增并且其中12个成员串联重复分布,从而使梅花具有独特的花香;推测梅花基因组中6个串联重复的DAM基因和其上游过多的CBF结合位点是梅花提早解除休眠的关键因子,从而解释“踏雪寻梅”之说。张启翔告诉记者,梅花全基因组测序的完成以及高密度遗传图谱构建,有助于揭示梅花花期早、花香独特等重要观赏性状的遗传基础,有助于挖掘与诸多重要性状相关的功能基因,为今后进一步揭示梅花花期、抗病调控机制、梅花及相关种属的分子育种奠定基础。研究中,项目组还揭示了蔷薇科植物进化规律。张启翔说,通过分析梅花的进化发现,梅与苹果发生分化后,并没有出现近期的全基因组复制事件,同时结合已完成的苹果和草莓基因组序列,成功重建了蔷薇科9条原始染色体,揭示了蔷薇科植物进化规律,为开展蔷薇科物种比较基因组学研究奠定重要的理论基础。据介绍,该科研成果由北京林业大学、深圳华大基因研究院及北京林福科源花卉有限公司等多家单位合作完成。目前,转录组数据组装及基因功能注释数据已在相关网站对外公开。
霍华休斯医学研究所,Baylor医学研究所的科学家们近期在PloS One上发表最新研究性文章,文章标题为:Big Genomes Facilitate the Comparative Identification of Regulatory Elements,该文章解析了基因组大小对基因组学的研究带来的影响。基因组越大则更容易找出控制基因活性的DNA区域。在小基因组上,功能性元件紧紧地结合在一起。而在大基因组上,功能性元件分得比较散,于是也更容易找到控制基因活性的区域。 基因组分为结构基因和调控基因,要从基因组上找到功能元件并不难,难的是找到调控基因表达的机制,因此,对小的基因组来说,紧凑的结构给寻找调控区域带领更多的困难,而相对来说大基因组却容易多了。功能元件散落在基因组上,更便于寻找调控区域。大的基因组更便于研究非编码DNA和RNA,对研究基因调控也更为有利。而目前,研究生命的遗传物质DNA的科学家一直觉得,基因组越小越受欢迎,因为操作简单,可以节省大量的时间和精力,尤其在金钱方面也能更节约成本,测序的费用更低。甚至有科学家说,基因组小则基因排列更紧凑,垃圾DNA也越少。 [img]http://www.instrument.com.cn/bbs/images/affix.gif[/img][url=http://www.instrument.com.cn/bbs/download.asp?ID=137848]Big Genomes Facilitate the Comparative Identification of Regulatory Elements[/url]
美国加州的山景城是“硅谷”的重要组成部分。现在,一个与硅芯片相关的潜力大产业正在这里兴起,那就是基因组测序技术产业。这个产业的发展是随着多家大公司的激烈竞争开始的。不过,一家名为“整合基因”(Complete Genomics,CG)的公司不像别的公司一样研发和销售测序仪器,而是为科学家提供外包的测序服务,更绝的是,在这家公司里做测序的,并不是研究人员,而是一排排的机器人。近日,《新科学家》杂志探秘了这家充满科幻意味的公司。前台都是“机器人”走进CG公司,连前台都由计算机终端出任。它会主动向来客问好,询问姓名、身份和来访意图。旁边连接的一台打印机则自动打出访客挂牌。与此同时,一份电子邮件已经发送到内部接应人员的电脑上。这家公司的生产线更像科幻电影里的实验室,昏暗蓝色的房间里到处都是高级仪器,室内温度保持在28℃和相对较高的湿度,几名穿着实验服,带着发罩的工作人员在监视着电脑屏幕,查看着机器人的运作状态。这儿已经成为了世界上最大的人类基因组测序工厂。只是在这里工作的不是人类,而是机器人。在一个大约只有半个网球场大的房间里,“坐着”16台机器人,不间断地进行着人类基因组测序的工作。去年,它们完成800个人的DNA测序工作其中三分之一是后半年做出来的。到了今年,它们已经可以每个月生产出400个人的基因了。CG公司只是目前迅速形成产业的诸多基因组测序公司中的一家,但是它十分独特。公司市场总监图柯特(Jennifer Turcotte)对《新科学家》杂志解释说,通常而言,DNA测序是在一个密封的机器里进行的,但在这家公司的实验室里,机器人却是在一个开放暴露的环境下做基因组测序,这是为了便于维修。实验室特定的温度和湿度是为了符合测序中出现的生化反应,微弱的蓝光是为了避免荧光探测剂在探测基因代码符号时受到其他频率光波的破坏。这儿所进行的基因组测序,已是目前最新的第三代基因组测序技术,称为“DNA纳米球测序技术”。这种新方法是将DNA链放置在一小块硅芯片上进行调节,自我组装成所谓的“纳米球”。这样的测序所需要的试剂更少,得到的数据则更多。技术人员都穿着无尘室服装,因为任何一点灰尘都会干扰测序,除非哪儿出问题了,一般而言这些技术人员不会干预机器人的工作。机器人则会自动添加试剂,操作样本,每个DNA纳米球上携带着70个核苷酸,其排列顺序会通过光信号被拍摄记录下来。费用正在逐步降低这些机器人正在做的工作,是一个浩大庞杂的工程蓝图中的第一步,所有的人类基因组中有着30亿对碱基对,而CG计划将其全部组装出来。这需要非常大的计算量,公司为此也建了一个自动数据中心。不过,这个数据中心设在距离公司大约有20分钟车程的地方那儿的电费更便宜。目前CG公司只针对研究者和制药公司开放,个人还没法购买他们的服务。在这里,每对基因组测序要价9500美元,如果购买1000对以上,则每对价格降为5000美元。这个价格是随着基因组测序技术突飞猛进而急剧下降的,要知道,十年前,第一对人类基因组序列完成时,其价格是以十几亿美元计量的。而科学家现在已经预计几年后,基因组测序的价格可能会降到一般人都可能支付得起的程度。基因组测序的流水线完全是由机器人来做的,而职员做什么呢?公司共有185名职员,部分是科研人员,忙于改善公司的测序技术,另一部分则是做市场和联络,与各类客户打交道。基因组测序工程是一项既有非常光明的前途但又异常庞大的科学工程,而自动化则可能成为处理这项工作的最佳工具。基因学家们认为,通过一些基因扫描,是可以找到导致人类易感疾病的一些基因变异,人类基因谱上,有一些常见明显变异,但是就整个遗传问题来看,还有大量的混乱的遗传变异隐藏在DNA双螺旋体中,这些也导致了世界上千奇百怪的遗传疾病。如何去捕猎这些神秘莫测的错误基因代码呢?只剩下一个方法,那就是将整个人类基因谱测序,来捕捉一些可能和疾病有关的基因变异。这个方法虽然听上去如同“大海捞针”一样不靠谱,但目前一些迹象表明,今后或许基因组序列会成为医疗记录的一部分,或者科学家可以通过家庭的基因组测序来纠正基因错误。比如,去年西雅图系统生物学研究所的胡德(Leroy Hood)及其小组与CG公司进行了合作,在《科学》杂志上刊登了一篇论文。他们对一家四口的基因组进行了测序。这是个特殊的家庭,两个孩子都患有两种隐性遗传病米勒综合征和纤毛运动障碍,而父母则完全正常,在分别测出这家人的基因序列后,研究者将父母和子女基因组序列进行比较,验证了米勒综合征这种非常罕见遗传病的致病突变。提供测序外包的服务目前,站在基因组测序产业化起跑线上的企业包括了同样位于加州的生物科学公司Pacific Bio。这个公司创立了首次可以对单个DNA进行测序的仪器。和CG公司一样,目前,这家公司也只向研究者提供服务。有一些大型的、从事基因组测序产业的公司已经将基因组测序做到医院和个人普及的地步了,如研发制造大型测序分析仪器的Illumina公司。这个公司在2008年美国成长最快的科技公司评选中,风头甚至盖过了Google。它们提供的产品甚至可以直接给病人使用。而另一位基因创业企业家罗斯伯格(Jonathan Rothberg)甚至发明了可以放在桌子上的基因解码器,可以在2小时之内以很高的精度解读出1000万个基因代码符号。大部分的基因组测序企业都站在一个竞争线上,尽力提高DNA测序的速度,降低费用。而CG公司其实并非和它们是严格意义上的竞争对手他们计划组装出所有的人类基因序列,研发也是为此目的而进行。此外,他们并不如其他公司一样开发更高级更小巧的基因组测序仪,而是为科学家提供基因组测序的外包服务,也就是说,研究人员无需购买、安装、培训、运行和维修仪器,而只要将样品交给这家公司,等待结果到来就可以。虽然很多人不理解他们的做法,但这家公司始终坚持自己的观点,认为这样的服务最能让科学家将时间从捣腾仪器设备的工作中解放出来,专心放在生物学和假说验证上。从这几年CG公司取得的成绩来看,这种做法确实是有效的。2009年,CG公司宣布其测出了第一个人类基因序列,并移交给美国生物科技信息中心数据库。同一年,他们在《科学》上刊文,发布了三个完整人类基因组序列分析的结果,当时文章还宣布,测序的成本已经可以降到1726美元。这在生物界引起了轰动。到了那一年结束,他们已经做出了50个人的基因序列。此外,他们的名字也随着来自各地的科学家一起多次登上了权威学术杂志。除了去年帮助科学家解开了米勒综合征突变难题给科学界留下难忘的印象之外,美国的罗氏公司还曾经借助CG的基因组测序技术,完成了人类科学史上第一例肺癌患者的全基因组比较。相关研究结果刊登在《自然》杂志上。而美国癌症学会也开始和CG公司联手,希望通过其服务比较正常人和癌细胞基因组序列的差异。或许在不久的将来,解开癌症之谜的第一个贡献就属于这些蓝光照耀下的机器人。
由中国科学家领衔的白菜、甘蓝和油菜全基因组测序项目取得阶段性重大成果,获得了白菜全基因组的精细图,甘蓝和油菜全基因组的框架图。 研究表明,白菜、甘蓝和油菜的基因组大小分别约为5亿、6.5亿和11亿个碱基对,白菜和甘蓝含有的基因总数目分别约4.2万和4.5万个,油菜基因覆盖度85%以上。该项成果是国际上首次对三个近缘作物物种进行的整体测序,并且油菜是迄今首个全基因组测序的异源四倍体植物,这不仅对研究作物进化和遗传改良有着重大意义,也对其他多倍体物种的全基因组测序具有重要的参考价值。 该项目分为白菜子项目和甘蓝、油菜子项目,前者由中国农业科学院蔬菜花卉研究所主持,参加单位有中国农业科学院油料作物研究所和深圳华大基因研究院,后者由中国农业科学院油料作物研究所主持,参加单位除上述两个单位外,还有国内湖南农业大学、西南大学、华中农业大学等和国外韩、英、加、澳、美等国家的相关研究机构。该项目得到了农业部、科技部以及国家自然科学基金委的大力支持。 白菜、甘蓝和油菜同属于芸薹属作物,油菜由白菜和甘蓝杂交后进化而来,它们的基因组分别命名为A、C和AC。白菜和甘蓝是我国主要的蔬菜作物,占全国蔬菜种植面积和产量的近五分之二 油菜是我国的主要油料作物之一,其食用油供给事关国家的食物安全。其全基因组序列测定将大大加速重要农艺性状控制基因的克隆和应用,从而给作物的产量、品质和抗病抗逆等重要农艺性状的改良提供基因资源和理论研究平台。
“10年前,我们参与人类基因组计划,完成了1%的工作,其实是‘搭了别人的车’。现在,面对即将到来的基因组学新时代,我们不能再搭别人的车了。”当年曾参与人类基因组计划承接1%测序工作、如今已是中科院北京基因组研究所副所长的[url=http://sourcedb.big.cas.cn/zw/zjrc/brjh/200907/t20090724_2194384.html][color=#800000]于军[/color][/url],对中国未来基因组学的发展和应用前途有点担心。2010年6月26日是人类基因组图谱公布10周年,国内外的一些研究机构都在这一天举行了纪念会。中国科协普及部、中科院北京基因组研究所、遗传与发育学研究所、中国遗传学会等单位也在北京举行了纪念会。会上,于军兴奋地回忆起当年他那个义无反顾的决定:1998年4月一天的早晨4点左右,他正在美国西雅图的家中睡觉。忽然自动传真机响了起来,“我爬起来一看,是邀请我回国工作的,我拿起笔签上自己的名字就传了回去”。这是于军回国工作的起点,也是中国参加人类基因组计划的起点。正是于军带回国内的技术和人才奠定了完成1%任务的基础。他是“1%计划”的“始作俑者”之一。与那时的热血沸腾相比,今天的于军更多了一份冷静与思考。“10年前,测定一个人的基因组,大约花了近10亿美元,用了13年的时间;而现在测一个人的基因组也就1万美元、一周左右的时间。美国现在已研制出第三代基因测序仪,用它测定一个人基因组的费用可降到1000甚至100美元,用时仅需15分钟。”于军说,当第三代基因测序仪广泛应用时,大规模应用基因组技术的“个体化基因组时代”就到来了。“个体化基因组时代”为人类描绘了一个美好的未来:那时,我们可以知道某一种药物为什么会对一部分人有治疗作用而对另一部分人不起作用,甚至起负作用;那时还会针对个体疾病的状态和遗传基础的独特性对症下药;也会针对个体化的药靶研制出个性化的治疗药物和治疗手段……这将是一个巨大的医疗市场。而测定每一个人的基因组本身也是一个大市场。“对于有十几亿人口的中国来说,假如使用美国研制的第三代基因测序仪来工作的话,那要进口多少台?按一个人测序需100美元计算,又要花费多少钱?”于军向记者言及此事,表现出了一种内心深处的忧虑。“中国一定要加快研制自己的DNA测序仪。”据了解,于军团队正与有关单位合作研制第二代和第三代测序仪器。“但我们的力量仍然有限,应该有更多的团队和单位加入到这个行列中来。这是我们迎接基因组学新时代的必要准备。”还有一种必要的准备,那就是做好有关基因、遗传学、基因组学等相关科学的科普宣传工作。“美国人十分重视基因组学的科普宣传。”于军回忆说,当年,美国立项测定人类基因组图谱时,就把这项工作的科普宣传列入了计划。10年前图谱完成时,时任美国总统克林顿发表致辞,电台、电视台现场直播,上万名美国人参加了当时各种各样的庆祝活动。“关于科普的作用,一个明显的例子是对待转基因食品的态度。”于军说,美国人就不像中国人那样对转基因食品“过分惶恐”。因为他们知道转基因食品并不像有人说的那样可怕和有危害。中国在面对转基因食品的问题上,好像是由大众的好恶来决定,而不是由对转基因的科学认识来决定。未来的基因组学时代、个性化基因组时代,我们可能会遇到比转基因食品更棘手的问题:法律问题、伦理道德问题、个人隐私问题等等。“从现在起我们就应该做好基因组新时代的科普宣传,未雨绸缪,为基因组学的发展和应用提供更加广阔的发展空间。”于军如是说。(转自科技日报)
由中美英等国科研机构发起的大型国际科研合作项目“千人基因组计划”10月28日在英国《自然》杂志上,以封面文章形式发布了迄今最详尽的人类基因多态性图谱,同时也在美国《科学》杂志上报告了在基因研究技术手段上的收获,相关成果标志着人类基因研究进入了一个划时代的新阶段。“千人基因组计划”由中国深圳华大基因研究院、美国国立人类基因组研究所、英国桑格研究所等机构于2008年启动,旨在绘制迄今最详尽、最有医学应用价值的人类基因多态性图谱。现在报告的是该计划第一阶段的分析成果。“千人基因组计划”共同主席、英国桑格研究所基因专家、《自然》封面文章主要作者之一理查德·德宾在接受记者采访时说:“这一计划现在取得了两个重要成果,第一是获得了迄今最详尽的人类基因多态性图谱,第二是探索出了研究基因多态性的新技术手段。”基因多态性是指人与人之间的基因差异。人的基因组总体上差不多,但在有些位置上你我他都不一样,存在各种基因变种,它们最终导致了人与人之间的差异。德宾说,在第一个成果方面,研究人员找出了1000多万个大大小小的基因变种,其中约800万个都是前所未知的。对于人群携带率在1%以上的基因变种,本次研究的覆盖率达到95%以上,得出了迄今最详尽的基因多态性图谱。这一成果在医学等领域有很高的应用价值,比如通过参照图谱,可以方便地找出致病的基因变种。在第二个成果方面,研究人员验证了在大型基因研究中综合使用多种基因测序手段的可行性。由于基因测序成本目前仍很高昂,如果能在“精测”一些基因序列的同时,对另一些基因序列只需“粗测”就能保证最终结果的准确性,将可以大幅降低基因测序研究的成本。《科学》杂志上的文章便侧重描述了技术手段方面的进展。德宾告诉记者,自十年前“人类基因组计划”完成以来,因为难以同时对许多人进行基因测序,基因研究一直只在较小的层面上进行。本次研究不仅使大规模测序成为可能,还绘制了一个详尽的基因图谱以供比对,这标志着人类基因研究进入了一个划时代的新阶段。他说,本次报告还只是基于“千人基因组计划”第一阶段中搜集的数百人的基因数据,而该计划的最终目标是获得欧、亚、美、非各洲不同人群中2500人的基因数据,预计在2012年发布的最终结果将可以覆盖99%以上的基因变种。据报道,“千人基因组计划”所获数据存放在公共数据库中,公众可免费查询。 (新华网)
历时6年,300余研究者花费5300万美金,牛的基因组序列终于呈现在世人面前,相关的文章发表在Science杂志上。这是继2000年人类基因组破解以来,又一动物基因组序列被破译。负责人称,牛的基因组的破译不仅有助人们更深入了解牛的驯化过程,提高牛肉,牛奶的质量改善人类的生活质量,还有助了解人类的疾病。最新的一期Science杂志刊登了两篇独立研究牛基因组的文章,一篇Genome-Wide Survey of SNP Variation Uncovers the Genetic Structure of Cattle Breeds;一篇The Bovine Genome Sequencing and Analysis Consortium,该项目对牛的基因组进行了分辨率精细的测序。另外还有一篇评论性的文章,The Genome Sequence of Taurine Cattle: A Window to Ruminant Biology and Evolution,将研究焦点放在对牲畜进化和驯养历史的追踪工作上。研究人员发现,牛的基因组含有至少2万2000个基因,其中大约有14345个基因在7种其它的哺乳动物种系中具有对应的基因。 这些发现显示,在牛的进化和驯养过程中,基因的数量和构成的变化是如何改变牛的生物学系统并对它们的繁殖、免疫能力、乳汁分泌和消化造成了最为显著的影响的。 这些研究人员还对来自19个不同地理和在生物学上混杂繁殖的497头不同牛只DNA中的3万7470种差异进行了调查。他们发现,母牛的进化与我们人类本身的进化截然不同,它们从一个有着非常大的有效祖先群体到近期发生的快速的群体下降,而不是反过来的那种一种情形。 文章的作者将这种进化归因于与以往驯化活动、因农业专门化所作的选择以及与动物豢养的形成相关的遗传学瓶颈。 但是,牛品种中的多样性的现有水平看来至少与那些在人类群体中的水平一样地强健有力。 在一篇Perspective中,Harris Lewin对这些发现进行了更为详细的探讨,并重点介绍了其对人类健康和可持续性农业的意义。
不过短短数十年,基因测序的价格从近30亿美元步入千元时代,这让你有没有恍如隔世的感觉?如果全基因组测序的价格降到非常低,你会选择测自己的基因序列吗?商家说做基因测序可以预知孩子有哪些天赋,你相信吗?Illumina、Life Tech、罗氏、华大基因,基因测序仪器市场谁主沉浮?中科院北京基因组研究所、中科院半导体所、北京大学、东南大学,学院派能否挑起测序仪器国产化的大梁?质谱技术也可以用来做基因测序,你听说过吗?虽然基因测序越来越容易,但我们得到的却是一本生命天书,要完全读懂它我们还需要多少时间?更多关于基因测序的信息,请访问:http://www.instrument.com.cn/news/subject/s325/
大熊猫基因组测序研究项目近日正式完成,并绘制出大熊猫基因组精细图。这是中国科学家第一次全面系统地对大熊猫基因组进行测序研究。 据介绍,大熊猫基因组测序研究结果表明,大熊猫有染色体21对,基因组大小2.4G,重复序列含量36%,基因2万多个。 这项研究由深圳华大基因研究院领衔,中国科学院昆明动物研究所、中国科学院动物研究所、成都大熊猫繁育研究基地和中国保护大熊猫研究中心共同参与。 研究结果还表明,大熊猫基因组仍然具备很高的杂合率和较高的遗传多态性;在已经进行全基因组测序的物种中,大熊猫基因组与狗的基因组最接近;数据分析结果同时还进一步支持了大多数科学家所持的“大熊猫是熊科的一个亚种”这种观点,证明了熊科内部各类群的分类情况。 据悉,大熊猫基因组精细图这一研究成果,填补了大熊猫基因组及分子生物学研究的空白,将从基因组学的层面上为大熊猫的保护、疾病监控及其人工繁殖提供科学依据。
近日,深圳华大基因研究院宣布,我国科学家将参与全球最大微生物基因组研究项目,对来自全球的20万个样本进行环境DNA测序或宏基因组测序,从而建立一个全球性的基因图谱,并承担核心工作。该项目旨在全方位、系统性研究全球范围内微生物群落功能及进化多样性,以便更好地造福社会及人类。与以往的微生物研究有所不同,该项目的研究对象不仅集中于海洋和人体环境中微生物群落,还包括土壤、空气、淡水生态系统等整个地球表面的绝大多数的微生物群落。华大基因将负责亚洲地区所有样本的收集和鉴定,并对整个项目提供DNA提取、扩增、建库、宏基因组测序以及研发生物信息学分析流程所需的计算资源。这些信息学分析流程将为项目研究产生的海量数据提供一个分析框架。项目负责人、芝加哥大学和阿贡国家实验室的教授杰克·吉尔伯特博士表示:“华大基因在测序能力、测序技术和信息分析等方面已展现出卓越的能力。此项目是一个前所未有的最大的基因组测序项目,作为全球最大基因组学研究中心,华大基因的参与至关重要。”华大基因理事长杨焕明院士表示,微生物对地球上所有的生命具有至关重要的作用,而我们对微生物的复杂性和多样性认识不足,征服这个未知的领域非常有必要。华大基因拥有国际先进水平的测序平台和强大的生物信息学分析能力,可以为促进人类对微生物群落重要性的了解贡献力量。(来源:科技日报)
由来自中国、美国、荷兰、以色列等14个国家的300多位科学家组成的“番茄基因组研究国际协作组”,历时8年多的艰苦努力,于近日完成了对栽培番茄全基因组的精细序列分析。今天,国际权威学术期刊《自然》以封面文章发表了这项重大科学成果。 番茄是研究果实发育的经典模式植物,我国科学家在这项国际番茄基因组研究中作出了重要贡献。作为中方协调人,中科院遗传与发育生物学研究所研究员李传友和薛勇彪负责第3号染色体的测序工作,中国农科院蔬菜花卉研究所研究员黄三文和杜永臣负责第11号染色体的测序工作。番茄基因组有12条染色体,中国科学家高质量地完成了番茄基因组测序总任务的1/6,标志着我国成为番茄基因组学研究的强国之一。 8年来,国际协作组采用“克隆连克隆”和“全基因组鸟枪法”相结合的测序策略,在解码的番茄基因组中,共鉴定出约34727个基因,其中97.4% (33840个)的基因已经精确定位到染色体上。番茄基因组的解读,是科学家通过国际合作完成的又一个高质量的模式植物的基因组序列分析,对于不同物种之间的比较基因组学研究具有重要价值,这项工作将极大推动番茄乃至包括马铃薯、辣椒、茄子等在内的茄科植物的功能基因组研究,为培育具有高产、优质、抗病虫害、抗逆等优良性状的番茄新品种打下了良好的基础,对推动全世界的番茄生产具有重要意义。 有关专家表示,我国蔬菜种业面临着强大的国际竞争。中国在国际蔬菜基因组研究领域具有优势地位,而如何把基础科研的优势转化为产业优势,是目前面临的主要挑战。科学家建议,应在进一步巩固蔬菜基因组研究优势的基础上,加强蔬菜作物分子设计育种体系的建设,并与常规育种相结合,加速有自主知识产权优良品种的培育,这对于支撑我国蔬菜产业可持续发展、提升我国蔬菜种业的国际竞争力具有重要意义,也是不可错过的历史机遇。
[font=宋体][size=3]中国医学科学院药用植物研究所与广药集团今天在京宣布“丹参基因组框架图”绘制完成。这是世界上首个药用植物基因组框架图。[/size][/font][font=宋体][size=3] 广州白云山和记黄埔中药有限公司与中国医学科学院药用植物研究所合作,利用第二代高通量测序技术对丹参全基因组进行测序,并完成丹参基因组框架图的组装。丹参基因组框架图的完成,对其它药用植物的研究具有很好的借鉴和示范作用,促进现代前沿生命科学研究和传统中药学的有机结合,将改变中药研究领域被动追赶其它学科发展的局面。[/size][/font]
科技日报讯 (记者王怡)中国科学院北京基因组研究所和吉林中科紫鑫科技有限公司4月18日在长春联合召开国产新一代基因测序仪样机观摩研讨会,向与会的国内基因测序领域专家和应用单位代表展示这一合作成果,同时向社会公开征集部分应用单位进行免费测试使用,测试工作将于今年下半年开展。 据了解,新一代基因测序仪样机是目前唯一技术性可以匹敌国际市场主流产品的国产基因测序系统,与国外第二代高通量测序系统相比,已经成功解决“读长较短”这一关键技术难题。该基因测序仪已经达到和部分超越国际主流设备技术指标,其成本低于进口设备1/3以上,应用成本低于进口设备1/5以上,使新一代测序技术真正达到进入广泛应用市场的经济条件,并将彻底解决我国基因测序仪完全依赖进口的局面。截至目前,该系统已获得7个发明专利和1个实用新型专利授权,还有多项专利正在申报。 中科院北京基因组研究所研究员于军介绍,新一代基因测序仪对传染性疾病的预防控制和诊疗,生物恐怖因子、食源性致病因子和转基因成分鉴定,口岸卫生和有害生物防御性检疫,以及针对人类遗传多样性而产生的疾病早期预警和个体化用药相关基因的检测分析等实践应用,提供强有力的技术支持。 据悉,测序仪今年下半年将在医疗、检验检疫、疾病防控、高校、研究院所等20家应用单位进行免费测试使用。已经确认参加测试应用的单位包括中国检验检疫科学研究院、北京出入境检验检疫局、青岛海洋大学等10余家单位。测试工作主要包括对系统性能的优化和改进,以及根据应用领域的不同进行应用产品的共同开发,即在基础试剂产品的平台上衍生出一系列专用型试剂产品,并开发相应的数据分析算法和数据库,实现测序技术实践应用的全面解决方案。来源:中国科技网-科技日报 2014年04月20日
最近,美国洛斯阿拉莫斯国家实验室(LANL)的一个遗传学小组和一国际财团联合提出了一套旨在阐明可公开获取的基因测序数据信息的质量标准。新标准最终可使遗传研究人员开发出更有效的疫苗,或有助于公共健康部门或安全人员更迅速地应对潜在的公共卫生突发事件。 在10月9日的《科学》杂志上,LANL遗传学家帕特里克钱恩和他的同事提出了6个基因组测序数据标签,可将基因测序数据按其完整性、准确性以及由此带来的可靠性进行归类。这些标签可在公共数据库中获取,而目前使用的标签仅为两个。此项成果的重要性在于,研究人员必须每天使用这样的数据,以对未知遗传数据和已知生物体的遗传数据进行相互参照,而有了这样的新的分类标准,数据的获取与对比工作的效率将大大提高。 每个生物体的细胞内都有DNA,由4个分子构建模块(或称碱基对)组成,碱基对排成特定序列时就可构成基因。这些基因序列可包含对生物体有益或有害的遗传指令。基因组研究人员编目了数以千计的基因数据,并将其放在公众数据库中以供其他研究者使用。然而,由于基因数据的复杂性,公共数据库中的遗传信息范围从粗略到精致一概都有。过去,这些基因数据常被归类为“草图”和“成品”两大类,给基因数据的准确性留下了太多的不确定性。 钱恩表示,在过去几年里,基因测序技术已取得重大进步,公众可获得的基因数据已呈爆炸性增长,每天产生的碱基对序列数据量要比过去几年产生的数据量还要多几十亿次。不同的测序技术具有不同的精确度。一个序列中的高度不确定性可能会引导研究人员走向一条耗时长达一年甚至数年的错误道路。因此,有必要建立一个标准,为研究人员提供对遗传测序数据质量的明确评估。 钱恩联合了大大小小的数个基因组测序中心,如美国能源部联合基因组研究所、桑格研究所、人类微生物群系项目Jumpstart联盟测序中心、密歇根州立大学以及安大略省癌症研究所等,共同提议将现有的测序数据分类从两大类充实为6大类。这6个标准涵盖了从代表公众提交最低要求的“标准草图序列”到代表最高标准的“完成序列”,而“完成序列”的验收标准是每10万个碱基对中最多只能包含一个错误。 LANL基因科学小组负责人、联合基因组研究所LANL研究中心主任克里斯戴特表示,该项研究的目的是为了让所有主要的基因组中心和基因组研究小组都能用上符合其需要的分类基因组测序数据。而为了尽可能保证基()因组序列的完整性,一些较小的研究中心也可采用这个分类等级来建立和提交其研究成果,以帮助其他科学家了解既已完成的工作。(科学网)
来自美国加州大学旧金山分校格拉斯通研究所的研究人员揭示出利用胚胎心脏细胞构建出完全功能性的心脏所需的上百个基因开关的精确开闭次序和时间。这项发现有助于对一些人先天性心脏病的遗传基础产生新的认识。在一项刊登于Cell期刊上的研究中,研究人员利用干细胞技术、下一代DNA测序和计算工具来将心脏细胞如何变成心脏的“基因组蓝图”拼接在一起。这些发现提供新的希望来对抗威胁生命的心脏缺陷,如心律不齐和室间隔缺损(ventricular septal defect)。在这项研究中,研究人员获取来自小鼠的胚胎干细胞,然后通过在盘碟中模拟胚胎发育而让它们分化为跳动的心脏细胞。接着,他们提取发育中的心脏细胞和成熟的心脏细胞内的DNA,并利用一种被称作ChIP-seq的高级基因测序技术来观察DNA中的表观遗传标记。论文共同第一作者Jeffrey Alexander说,“但是发现这些标记只是成功的一半,因此我们接着不得不破解它们编码心脏形成的哪些方面。为此,我们利用格拉斯通研究所生物信息学核心(Gladstone Bioinformatics Core)的计算能力。这允许我们获得基因测序中所收集的大量数据,并且将这些数据组装成一种可读的和有意义的将心脏细胞如何变成心脏的蓝图。”研究人员获得了一些意料之外的发现。他们发现在心脏细胞中,一组基因似乎以一种协作的方式一起发挥作用:在胚胎发育的指定时间,这组基因一起开启和关闭。他们不仅鉴定出很多参与心脏形成的新基因,而且也精确地确定地这些新发现的基因如何与之前已知的基因相互作用。绘制出心脏的基因组蓝图对人类健康的影响非常深远。鉴于研究人员理解这些基因如何控制心脏形成,他们能够开始将心脏病如何破坏这种调节的细节汇聚在一起。最终,他们能够寻找疗法来阻止、中断或抵消患有先天性心脏病的儿童体内这种调节遭到的破坏。
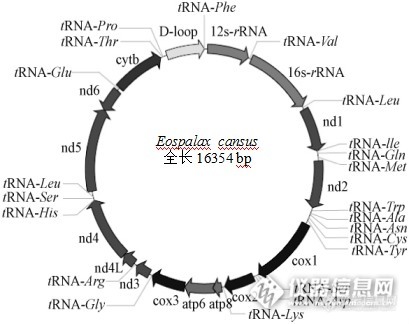
[b][/b][align=center]鼢鼠([i]Eospalax[/i])线粒体基因组测定及注释分析[/align][align=center]西安国联质量检测技术股份有限公司[/align][align=center]安平中心:李瑞[/align][b]摘要【[/b]目的】获得鼢鼠线粒体基因组全序列,为线粒体基因组功能标记及进化生物学等研究提供基础资料。【方法】参考鼹型鼠等动物的线粒体基因组序列,设计出可覆盖鼢鼠线粒体基因组的16对引物,采用[url=https://insevent.instrument.com.cn/t/jp][color=#3333ff]PCR[/color][/url]产物直接测序法测得甘肃鼢鼠线粒体基因组全序列,分析其基因组的特点和基因结构。并结合GenBank中发表的啮齿类动物基因组全序列,探讨啮齿类动物的系统进化关系。【结果】鼢鼠线粒体基因组全长16354bp,其中包括22个tRNA基因、13个蛋白质编码基因、2个rRNA基因和2个D-loop区。碱基组成为33.5%A、24.2 %C、12.3 %G、30.0 %T。【结论】鼢鼠线粒体基因组结构及其信息和其他啮齿类动物的结构一致,线粒体变异保守。研究结果为鼢鼠的低氧适应、系统发育关系等提供了基础资料。[b]关键词 [/b]鼢鼠;线粒体基因组;序列分析 鼢鼠([i]Eospalax[/i])是分布于我国的主要啮齿类动物之一,其体型较小,栖息于洞穴内有挖掘活动,扩散能力强,数量波动大,是生态系统中重要的初级消费者,处于生态系统中的中心位置,草原生态系统中其能流比重很大[sup][/sup]。动物线粒体([color=#333333]Mitochondrion[/color])基因组为双链闭合环状分子[sup][/sup],少数也有线性的,它们具有分子量相对较小、结构简单、缺少重组、母性遗传和进化速率快等特点,已成为动物系统发育与进化、群体遗传学、分子生态学以及疾病机理研究等领域的理想材料[sup][/sup]。甘肃鼢鼠是仅分布于我国西北部的土著物种,其外形似中华鼢鼠,主要分布于甘肃临潭县及其附近地区。目前对线粒体DNA的研究主要在动物分子遗传学、分子生态学、种群遗传结构分析、遗传多样性、物种和品系鉴定、保护遗传学等方面得到了广泛应用[sup][4[/sup][sup],[/sup][sup]5][/sup]1. [b]实验材料和方法[/b]1.1 实验材料鼢鼠:采集于天祝(经度102.84、纬度 37.2)1个群体;鼢鼠解剖采集肝脏及肌肉组织样品,-20℃保存备用。1.2 线粒体DNA的提取用剪刀将肝脏及肌肉材料剪成小块,取0.1cm左右的小块肝脏及肌肉材料,采用常规的SDS/蛋白酶K裂解,酚氯仿提取DNA[sup][/sup],使用琼脂糖凝胶电泳检测其完整性。1.3 引物设计和[url=https://insevent.instrument.com.cn/t/jp][color=#3333ff]PCR[/color][/url]扩增通过Clustal X1.83比对,寻找相对应保守区域位置,用Primer Premier5.0引物设计软件设计引物,并对每条引物进行评价和修改,最终确定16对引物。以所提取的DNA为模板,用16对引物扩增覆盖整个线粒体基因组。利用引物进行[url=https://insevent.instrument.com.cn/t/jp][color=#3333ff]PCR[/color][/url]扩增,反应体系总体积为50μL,其中含有6μL [url=https://insevent.instrument.com.cn/t/jp][color=#3333ff]PCR[/color][/url] buffer、3μL MgCl[sub]2[/sub](1.5mmol)、MgCl[sub]2[/sub],2μL dNTPs (100μL mol)、上下游引物各2μL (0.25μL mol)、Taq DNA聚合酶2μL (1U)、总DNA约为2μL (25ng)、去离子水31μL。反应程序为:94℃预变性4 min,94℃变性50s,48-45℃退1min,72℃延伸1 min 30s,循环30次,之后72℃延伸10min,并根据不同引物的退火温度和扩增反应的实际效果进行优化。取 5 μL [url=https://insevent.instrument.com.cn/t/jp][color=#3333ff]PCR[/color][/url]扩增产物,和2 μL DNA marker 2000,进行1.0%琼脂糖凝胶(1×TBE)5V/电泳,用紫外观察[url=https://insevent.instrument.com.cn/t/jp][color=#3333ff]PCR[/color][/url]产物扩增情况,凝胶成像仪扫描记录结果。1.4 纯化、测序和序列拼接 在[url=https://insevent.instrument.com.cn/t/jp][color=#3333ff]PCR[/color][/url]产物中加入5 U SAP和2 U ExoⅠ,震荡混匀,37℃保温1 h,然后75℃保温15 min以灭活SAP和ExoⅠ酶,纯化好的模板可以在4℃保存24 h或-20℃长期保存。将纯化后的引物送往上海生工生物技术服务有限公司用ABI-3730序列自动分析仪进行双向测序。利用DNASTAR和测序峰图结果分析软件Chromas 2.22校对测序图,DNAMAN拼接序列。得到甘肃鼢鼠线粒体全基因组全序列。2. [b]结果[/b]2.1 鼢鼠线粒体基因组基因定位2.2.1 鼢鼠线粒体2个rRNA的分析哺乳动物线粒体的rRNA具有高度的保守性,它们的位置固定,12S rRNA位于tRNA-phe 和tRNA-Val之间,16S rRNA位于tRNA-Val和 tRNA-Leu之间,12S rRNA起始位置为68,终止位置为1019,长度为952bp,16S rRNA起始位置为1086,终止位置为2651,长度为1566。同时我们比对了鼢鼠和中华鼢鼠的rRNA基因和蛋白质基因,12S rRNA和16S rRNA的相似性分别为91.0%和87.3%,高于蛋白质编码基因之间的相似性。2.2.3 鼢鼠线粒体基因组结构 除NADH脱氢酶亚基6外均在H链上,虽然鼢鼠染色体数目少、染色体大,但与其它哺乳动物线粒体全基因组相比,它的线粒体基因组的结构与其它哺乳动物是十分相似的。甘肃鼢鼠线粒体基因组结构见图1。[align=center][img=,409,324]http://ng1.17img.cn/bbsfiles/images/2017/09/201709081454_02_2904018_3.png[/img][/align]注:ND: NADH脱氢酶亚基(NADH dehydrogenase subunit)、Cox:细胞色素氧化酶亚基(cytochrome oxidase subunit)、Atp:ATP合成酶亚基(ATP synthase F0 subunit)、Cyt b:1个细胞色素b编码基因(cytochrome b)。[align=center][b]图1[/b] 甘肃鼢鼠线粒体基因组结构简图[/align][align=center]Fig.1 The gene organization of [i]Eospalax cansus[/i] mitochondrial genome[/align]3. [b] 讨论[/b] 甘肃鼢鼠线粒体基因组的D-loop区,长度为933bp,比中国地鼠D-loop区(867bp)长。D-loop区对目的基因是不可缺少的,虽然D-loop区不能编码蛋白质但对于遗传信息表达是不可缺少的,在它上面有调控遗传信息表达的核苷酸序列,具有遗传效应的,比如RNA聚合酶结合位点是具有遗传效应的。8只甘肃鼢鼠中有5个单倍型:3只临潭群体共享1个单倍型,2只天祝群体独享单倍型;其余个体均独享单倍型,表明了甘肃鼢鼠线粒体DNA D-loop区碱基变异快、进化快的特性,符合啮齿动物线粒体变异大的现象。随着研究的深入,以线粒体DNA中完整的基因序列或多个基因序列协同而获得遗传信息来探讨物种的系统进化关系,将是以后研究发展的主要方向[sup][/sup]。目前,线粒体DNA已经在许多哺乳类动物的起源进化的研究中取的了重大进展,而对甘肃鼢鼠的起源进化的研究却很少,并且存在着甘肃鼢鼠属于[url=http://baike.baidu.com/view/113192.htm][color=#000000]瞎鼠科[/color][/url]和仓鼠二者之争,因此,为了更好的阐明甘肃鼢鼠的起源,还需要做更多、更深入的研究。
新的遗传学技术揭示了肥胖基因的变异编译 zfyyzz00字数544 《科学日报.》在2010年11月29日报道 - 肥胖是高度遗传的,但到目前为止遗传关联研究只揭示了这种遗传本质一小部分。现在,在生物医学可以公开获取的杂志《Genome Biology》上的一项研究中,研究人员在两个神经系统基因已经证实了DNA的,它们与过高的与体重指数相关。来自美国加州大学圣地亚哥分校和斯克里普斯转化科学机构的Kelly Frazer和同事以及Sanofi - Aventis使用了一种新方法,它可能在寻找隐藏的遗传特性成为普及:在一个大量人群中重新对基因组的候选区域进行测序,然后再与该疾病有关基因区域寻找遗传标志物。Frazer说:“我们测序编码酶FAAH和MGLL的两个区间,它们是调节在大脑和周围组织内源性大麻素水平,参与能量平衡和调节食欲。这些内源性大麻素水平在参与的肥胖患者中具有高水平,这两种酶从而提供了强有力的候选基因,来解释与体重指数相关的遗传特性”。在这两个基因,研究人员能够确定四个区域与BMI相关:FAAH启动子,MGLL启动子子,MGLL内含子2和MGLL增强子。这些区域的另一项测试显示,在血浆中存在内源性大麻素水平升高有关的罕见变异,这与以往的研究结果一致。Frazer说:““这是使用新的测序技术首次研究,把诸如肥胖少见的低频变异与复杂的通路相联系,并将特别关注的是要了解更全面的肥胖遗传性的作用,一这是一个在全球日益严重严重的健康问题。”编者按:本文并非旨在提供医疗咨询,诊断或治疗。出处:http://www.sciencedaily.com/releases/2010/11/101129203332.htm
人类基因组单核苷酸多态性的研究进展与动态The research development of single nucleotide polymorphisms in human genome 摘要:第一张人类基因组序列草图已经公布,正式图预计也将于2003年4月完成。但序列图只基于少数个体,它反映了基因组稳定的一面,并未反映其变异或多态的一面,而正是这种多态性,即基因组序列的差异构成了不同个体与群体对疾病的易感性、对药物与环境因子不同反应的遗传学基础。人类基因组中存在广泛的多态性,最简单的多态形式是发生在基因组中的单个核苷酸的替代,即单核苷酸多态性(single nucleotide polymorphisms, SNPs)。SNP通常是一种二等位基因的(biallelic),即二态的遗传变异,在CG序列上出现最为频繁。在转录序列上的SNP称为cSNP。SNP的数量大、分布广。按照1%的频率估计,在人类基因组中每100~300个核苷酸就有一个SNP。因此,整个人类基因组(3.2 X 109bp)中至少有1,100万以上的SNPs,在任何已知或未知基因内和附近都可能找到数量不等的SNP 目前普遍认为,作为数量最多且易于批量检测的多态标记,SNP在连锁分析与基因定位,包括复杂疾病的基因定位、关联分析、个体和群体对环境致病因子与药物的易感性研究中将发挥愈来愈重要的作用。迄今,对多基因疾病候选基因的SNPs研究已积累了丰富的数据,基于这些SNPs的关联分析也正方兴未艾。本文阐述了SNP的特征、不同研究者对基于SNP进行关联分析的观点以及SNP的研究进展与动态。 关键词: SNP;遗传标记;关联研究 中图分类号:Q75 随着分子遗传学的进展,疾病遗传学研究从简单的单基因疾病转向于复杂的多基因疾病(如骨质疏松症、糖尿病、心血管疾病、精神性紊乱、各种肿瘤等)与药物基因组学的研究中。与前者相比,多基因性状或遗传病的形成,受许多对微效加性基因作用,即其中每种基因的作用相对较微弱。这些不同基因构成的遗传背景中,可能有易感性主基因(major gene)起着重要作用。它们同时还受环境因素的制约,彼此间相互作用错综复杂,所以任一基因的多态性对疾病发生仅起微弱的作用。鉴于此,需要在人类基因组中找到一种数目多、分布广泛且相对稳定的遗传标记,单核苷酸多态性(single nucleotide polymorphisms, SNPs)正是代表了这样一种标记,所以它成为继第一代限制性片段长度的多态性标记、第二代微卫星即简单的串联重复标记后,第三代基因遗传标记。 1. SNP作为遗传标记的优势 SNP自身的特性决定了它比其它两类多态标记更适合于对复杂性状与疾病的遗传解剖以及基于群体的基因识别等方面的研究。 (1)SNP数量多,分布广泛。据估计,人类基因组中每1000个核苷酸就有一个SNP,人类30亿碱基中共有300万以上的SNPs。SNP 遍布于整个人类基因组中,根据SNP在基因中的位置,可分为基因编码区SNPs(Coding-region SNPs,cSNPs)、基因周边SNPs(Perigenic SNPs,pSNPs)以及基因间SNPs(Intergenic SNPs,iSNPs)等三类。 (2)SNP适于快速、规模化筛查。组成DNA的碱基虽然有4种,但SNP一般只有两种碱基组成,所以它是一种二态的标记,即二等位基因(biallelic)。 由于SNP的二态性,非此即彼,在基因组筛选中SNPs往往只需+/-的分析,而不用分析片段的长度,这就利于发展自动化技术筛选或检测SNPs。主要的技术方法包括单链构象多态性(single strand conformation polymorphisms, SSCPs)法、异源双链分析(heteroduplex analysis, HA)、DNA直接测序分析、变异检测阵列(variant detector arrays, VDA)法以及基质辅助激光解吸附电离飞行时间(MALDI-TOF)质谱法等。 (3)SNP等位基因频率的容易估计。采用混和样本估算等位基因的频率是种高效快速的策略。该策略的原理是:首先选择参考样本制作标准曲线,然后将待测的混和样本与标准曲线进行比较,根据所得信号的比例确定混和样本中各种等位基因的频率。 (4)易于基因分型。SNPs 的二态性,也有利于对其进行基因分型。对SNP进行基因分型包括三方面的内容:(1)鉴别基因型所采用的化学反应,常用的技术手段包括:DNA分子杂交、引物延伸、等位基因特异的寡核苷酸连接反应、侧翼探针切割反应以及基于这些方法的变通技术;(2)完成这些化学反应所采用的模式,包括液相反应、固相支持物上进行的反应以及二者皆有的反应。(3)化学反应结束后,需要应用生物技术系统检测反应结果。目前许多生物技术公司发展出高通量检测SNP的技术系统,如荧光微阵列系统(Affymetrix)、荧光磁珠技术(Luminex,Illumina, Q-dot)、自动酶联免疫(ELISA)试验(Orchid Biocomputer)、焦磷酸的荧光检测(Pyrosequencing)、荧光共振能量转移(FRET)(Third Wave Technologies)以及质谱检测技术(Rapigene, Sequenom)。 2. 基于SNP的关联研究 如果某一因素可增加某种疾病的发生风险,即与正常对照人群相比,该因素在疾病人群中的频率较高,此时就认为该因素与疾病相关联。如非遗传因素吸烟与肺癌相关;在遗传因素中,如APOE4与Alzheimer`s相关。对疾病进行关联分析需要在年龄与种族相匹配的患者和对照人群中确定待测因素(环境的或遗传的)的频率分布,患者和对照人群的选择是否恰当直接影响结果的可靠性。对常见的由高频率、低风险等位基因导致的疾病,采用致病等位基因的关联分析比连锁分析更有效。 应用SNP进行关联研究,首先需明确多少SNPs才可满足在全基因组范围内的分析。Kruglyak应用计算机模拟法预测人类基因组中超过3Kb就不存在连锁不平衡,据此推出完成全基因组扫描将需要500,000个SNPs。而Collins等收集通过家系研究得到的常染色体单倍型的信息发现,在染色体上相距0.2cM到0.4cM(约200-400kb)之间的标记仍存在连锁不平衡,如按每100kb需要一个SNP计算,那么完成全基因组扫描仅需约30,000个SNPs,平均每3-4个基因用一个SNP就可识别出整个基因组内任何位置上的具表型活性的变异。最近发现SNP与SNP之间的连锁不平衡甚至可延伸到更远的区域(0.35cM-0.45cM),那么进行基因组扫描需要的SNP数量就更少。导致上述估算SNP 数量差异的主要原因是Kruglyak进行模拟计算时,假设现在的人群在5000年前起源于共同的祖先,且人群规模的有效大小保持在10,000左右,然后经过连续的指数扩增,直至达到现在的50亿左右。Collins认为这种假设是不现实的,在人类发展的历史过程中,人群数目的增长是迂回曲折的,经历扩张与萎缩的周期性变化。 Weiss等认为Collins及其同事的结果可能低估了问题的复杂性。因为他们的结果或是基于小样本资料推断出来的,就会使连锁不平衡(LD)程度的估算偏高;或是从理论上预测LD的水平,而忽略了基因组中大量的随机变异。如大多数位点的信息是来源于小样本中测序得到的资料,据此得到的单倍型结构不可靠。目前的研究集中于基因组中LD相对广泛存在的区域,在此区域内,基因相对容易作图。如基于这些经验来进行基因组其它区域的LD分析,就可能发生偏离。如两个相距较远的SNPs 之间具有强的LD性质,就认为它们之间的SNPs及该SNP侧翼的SNPs也存在强烈的LD,这种假设仅适合于其中一些多态位点,但它并不是通则。当然,在一些罕见人群中,如Saami,在较长的区域内广泛存在大量的LD,但对Fihland人群,则在较长区域内几乎不存在LD,对全球整个复杂人群而言,LD肯定变得更复杂一些。 Gray等认为随着人类基因组测序计划的进展,人类基因组的结构逐渐被阐明,因此就可在那些富含基因的区域选择SNP进行全基因组扫描,这样所需的SNP数量还会减少。Halushka等根据他们对75个基因检测的实验结果推测,SNPs在单个基因或整个基因组中的分布是不均匀的,在非转录序列中要多于转录序列,而且在转录区也是非同义突变的频率比其它方式突变的频率低得多。Templeton 等对LPL基因突变与重组热点的研究结果提示,SNP集中分布于基因组的CG二核苷酸处或单核苷酸重复区或αDNA聚合酶的识别位点(TGGA)处。将人类基因组不同区域物理图谱与遗传图谱的进行比较,发现遗传距离和物理距离的比值有很大的差异,提示基因组不同区域的重组水平存在差异。如Dunham等将22号染色体STR的物理位置与遗传位置进行了对比,发现该染色体的重组率差异很大,提示存在重组热点。根据基因组内不同区域重组频率的高低可进一步选择SNP的数量,重组热点需要的标记数量就多,相反就少。这种设计也可能会进一步减少基因组扫描所需的SNP标记。 使用SNP进行关联分析面临的另一个问题是如何选择SNP。如果对每一个SNP都进行独立研究,那么对几百万SNPs 的研究就会导致成千上万次的假关联,结果就掩盖真实的关联性,所以,进行关联分析前,一定要对所研究的SNP进行选
国家“863计划”现代农业技术领域在主要动植物功能基因组研究方面,利用“十五”建立的水稻功能基因组的技术平台,系统开展水稻产量、品质、抗病抗逆、营养高效性状的功能基因组研究,克隆验证新基因和调控因子,应用芯片技术建立水稻重要农艺性状的全基因组表达谱,并开展比较基因组学研究和第3、4染色体功能基因的系统鉴定。 利用水稻、拟南芥等模式植物功能基因组的技术平台,开展小麦、玉米、棉花、油菜、大豆、花生、番茄等作物的功能基因组研究,克隆验证重要农艺性状基因;建立家蚕和家鸡的功能基因组研究技术平台,分离克隆与家蚕丝蛋白质合成、性别决定、发育变态、分子免疫和对微生物抵抗性、鸡的生长、品质、抗性、繁殖等重要经济性状相关的重要功能基因和调控因子。







 我要推广仪器
我要推广仪器
 下载APP
下载APP