[color=#444444]如果知道一种物质的结构,大家一般是通过什么途径搜索到相应的或者相近结构物质的液相色谱条件的文献的呢,在自己摸索之前总想找点参考,可是一直没接触过试条件,望各位传授一下文献搜索的路径啊[/color]
[color=#444444]我现在检测的是头孢类药物中的一个中间体 极性很大 类似盐 想用反相液相色谱法检测纯度 没有太多经验 找不到好的液相条件 还请老师们告诉我这类化合物怎么摸索条件 万分感谢 ![/color]
[color=#444444]最近合成了一个新化合物,是紫杉醇和一个小分子羧酸生成的酯,分子量1042。这个新化合物没人报道过怎样用液相测含量,紫杉醇含量测定大都是乙腈:水(1:1,V/V)。我的这个新化合物可以完全溶解在乙腈或者甲醇中。我用紫外扫过这个化合物最大吸收波长,发现它跟紫杉醇最大吸收波长几乎一样。请问我应该摸索这个化合物的色谱流动相条件,是用纯乙腈开始冲吗?非常感谢!![/color]
要做个液相色谱,但是像流动相,流速,检测波长什么的条件需要自己设定,有谁自己摸索过的,可以给我讲讲怎么弄的吗?~
当你在使用梯度进行方法摸索之前,你会做哪些准备工作?低压梯度和高压梯度对方法摸索的影响及局部分离不理想时应该如何改进?有关三元梯度洗脱的技术与文章,不胜感激!
各位同行有做二元脂肪酸液相色谱法定量的吗?基本是什么柱子比较合适?
本学生即将工作,重点在于操作液相[url=https://insevent.instrument.com.cn/t/Mp]气相[/url],我想知道拿到一药品,该怎么样去摸索液相的流动相,因缺少经验,还望各位朋友帮帮忙.谢谢!

“色”路蹒跚,即鹿无虞,某品种有关物质方法学流动相摸索。因暂时没有CERI L-column色谱柱,挑选了以前常用的色谱柱,进行了流动相的摸索,日产某品牌因理论板数较低(小于2000),故没有采用,选用月旭的色谱柱,一切皆好,试验完成后,把工艺处方摸索的样品都测试完成后,发现是用的苯基柱,虽说都是反相色谱柱,和标准不一致,要验证的,真是即鹿无虞啊!有关物质避光操作。照高效液相色谱法(中国药典2010年版二部附录Ⅴ D)测定。色谱条件与系统适用性试验 用十八烷基硅烷键合硅胶为填充剂(CERI L-column ODS,4.6mm×250mm,5μm或同等效能的色谱柱);流动相A为pH3.8醋酸钠缓冲液,流动相B为乙腈,按下表进行梯度洗脱: 时间(分钟)02040707180流动相A(%)606030306060流动相B(%)404070704040柱温为40℃,检测波长245nm。取苯甲酸乙酯约0.2g,置50ml量瓶中,加乙腈-水(3:2)溶解并稀释至刻度,混匀。取此溶液和供试品溶液各5ml置同一50ml量瓶中,用乙腈-水(3:2)稀释至刻度,摇匀,作为分离度测试溶液,量取10μl注入液相色谱仪,调整色谱系统,使匹伐他汀的保留时间约为23分钟,取对照溶液重复进样6次,主峰面积的相对保留时间偏差应小于9%,匹伐他汀与苯甲酸乙酯峰之间的分离度不小于5。测定法 取本品5片,置50ml量瓶中,加水20ml,振摇10分钟至片剂崩解,再超声5分钟,加乙腈20ml,振摇10分钟,加乙腈稀释至刻度,摇匀,滤过,取续滤液作为供试品溶液;精密量取供试品溶液适量,加乙腈-水(3:2)稀释制成每1ml中约含匹伐他汀钙0.2μg的溶液,作为对照溶液。量取对照溶液10μl,注入液相色谱仪,调节检测灵敏度,使匹伐他汀的峰高约为记录仪器量程的5~10%,再精密量取供试品和对照溶液各10μl,注入液相色谱仪,记录色谱图。供试品溶液色谱图中除溶剂峰与辅料峰外,差向异构体(相对保留时间约为1.1)、内酯化合物(相对保留时间为1.7)和其他未知杂质的峰面积分别不得过对照溶液主峰面积的7.5倍(0.75%)、5倍(0.5%)和1倍(0.1%),杂质总量不得过对照溶液主峰面积的20倍(2.0%)。色谱柱信息:1.日产的C18色谱柱(都是大家最常用和常见品牌,未写出,主要怕麻烦)。2.月旭的C18色谱柱,在使用的过程中,操作人员弄成月旭的苯基柱。苯基柱信息:苯基柱:SN:241301633, LN:2401.11 日产C18色谱图:http://ng1.17img.cn/bbsfiles/images/2013/07/201307132344_451180_1621890_3.png上图显示,理论板数较低,只有1834,虽说都是新柱子,也可能是流动相不适合该色谱柱,保留时间较短,要达到23分钟,调整流动相比例,估计理论板数会更低,故没有再折腾。http://ng1.17img.cn/bbsfiles/images/2013/07/201307132348_451181_1621890_3.png上图显示,其基线重现性良好,显示了新柱子的基本性能。月旭苯基柱色谱图:http://ng1.17img.cn/bbsfiles/images/2013/07/201307132349_451182_1621890_3.png上图显示,理论板数较高,超过了10000,分离度和拖尾因子均较好,调整流动相比例,应该是有可能的。调整流动相比例色谱图:http://ng1.17img.cn/bbsfiles/images/2013/07/201307132350_451183_1621890_3.png上图显示,分离度,拖尾因子,理论板数均好,大功告成(高兴的较早哈,呵呵。。。)下图为放大显示:http://ng1.17img.cn/bbsfiles/images/2013/07/201307132352_451184_1621890_3.png总结,做试验是个细致活,在没有充足的条件下,创造条件,是好事,但是要注意细节(注意看标签,不要看到月旭的就想到了是C18),不然会走弯路的,我现在要进行色谱柱验证了,和C18进行对比。
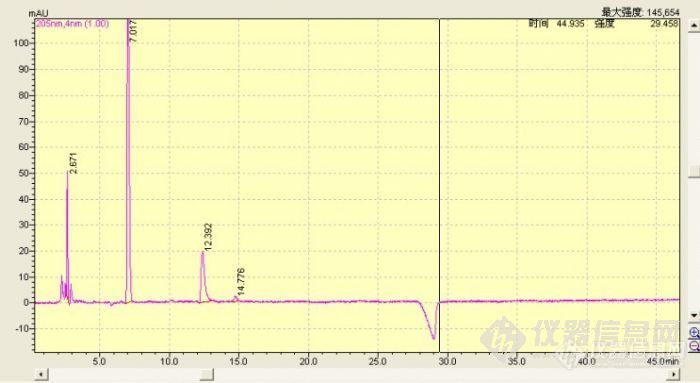
液相色谱检测条件选择问题?? 现在做一个样品,液相色谱条件是:0.05M 磷酸二氢钾-乙腈,用的是氨基柱。结果样品不纯,有好多杂质峰,现在想去做质谱,看看是什么杂质,可是质谱是不能用磷酸盐的,所以得再摸索一个条件。在液相上摸索的质谱条件是:乙酸铵溶液-乙腈,用的氨基柱。可是结果与之前的液相条件对比,杂质峰的个数少了,这样去做质谱也没意义啊?求助下这种情况质谱的条件该怎么选啊?http://ng1.17img.cn/bbsfiles/images/2010/12/201012131422_266661_2163534_3.jpghttp://ng1.17img.cn/bbsfiles/images/2010/12/201012131422_266662_2163534_3.jpg
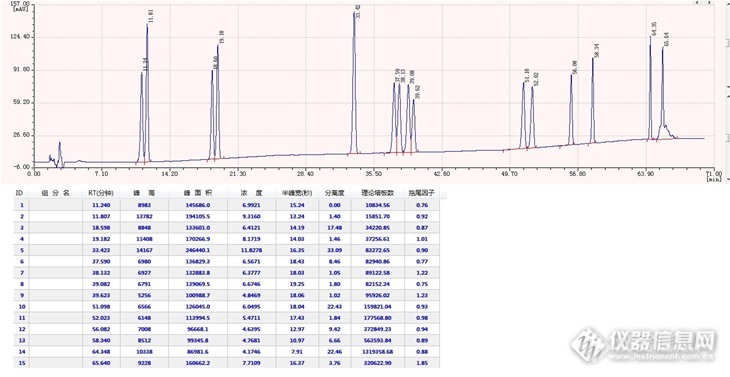
16种塑化剂的液相色谱方法开发终于算是完成了,整整一个月的时间,换过不同的柱子,尝试过各种不同的流动相种类和配比,250多次方法摸索和进样,一针一个多小时啊,项目逼得紧,周末无休,简直都快疯掉了。这16种塑化剂同时分离太难了,好不容易把这两个挨得很近的化合物分开,那两个又并一块儿去了,特别是第三组的四个峰DIBP、BBP、DBP和DBEP,真的是按下葫芦浮起瓢,如果流动相中乙腈多一点前两个峰分得开,可后两个峰却粘得更紧了,而甲醇多一点则前两个峰又粘一块儿去了,而且水的多少也是一个不可忽视的变量。由于是同时分析,所以每个峰必须都要能看得到,头大啊,一个月中大概有3/4的时间都是在解决这四个峰的分离度问题,最后把流动相调成A相是水:乙腈=60:40、B相是甲醇:乙腈=60:40的情况下才实现了这四个峰各自1.0以上的分离度。邻苯二甲酸二辛基酯和二壬基酯在C18上根本分不开,选择性不行,在C8上却很好的实现了基线分离,看来C8和辛基(也是8个碳原子)确实体现了比较好的选择性的一面。以下是谱图和数据,与大家一起分享。液相色谱仪:WUFENG LC100 二元高压梯度系统色谱柱:PntulipsTM BP-C18, 5um, 4.6×250mm流动相:梯度时间(min)A相:水/乙腈=60/40B相:甲醇/乙腈=60/4001000310002150[align=cent
大家好我是大二的学生,我们学校有高效液相色谱-质谱联用经过一年多的接触和摸索。我发现这些仪器都有些不尽人意的地方(比如有的仪器里的面板在连接处有缝隙容易掉进东西,有的手动螺母不好拧动,管路等)。一直以来我都在想一些可行的改进方法。我今后的目标是设计出以自己命名的仪器。所以在这里,我想搜集些大家的意见,您用的是什么仪器,在哪些方面您觉得不方便,需要改进,请回复我谢谢!
手头的液相色谱柱与资料上查的柱子规格(比如柱长,柱内径,液膜厚)不同,尤其是在梯度洗脱时,如何运用资料上提供的色谱条件,在自己实验室摸索出流动相条件。特向各位同人请教。
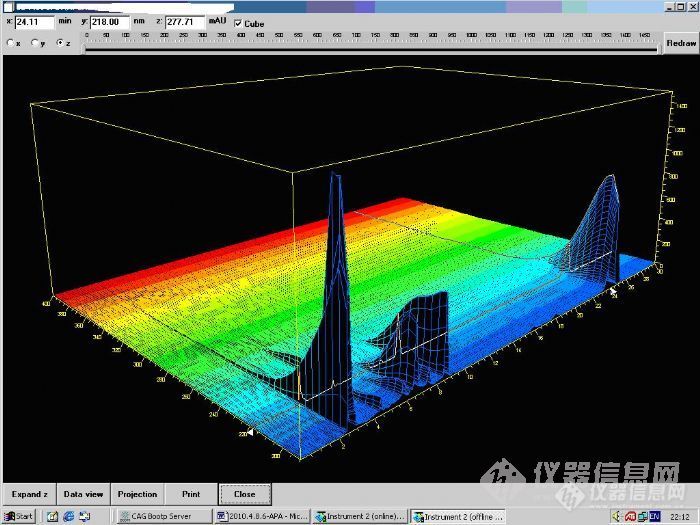
本人正在做一个合成,酰化反应,用高效液相色谱法来判定反应的终点。由于本人底子差,不懂如何做,我查了资料,但是对具体的关键步骤基本都没有,现将我对于液相条件的摸索,希望大家指点指点。由于反应底物和产物老师需要保密,所以我就暂时不告诉大家。柱子:月旭柱,XB-C18,5μm,4.6*250mm。1、这是底物峰。液相条件:月旭C18柱,柱温25,流动相,乙腈:磷酸盐ph8,25mmol=10:90.流速1ml/min。检测波长220nm。http://ng1.17img.cn/bbsfiles/images/2010/04/201004182319_213083_1605610_3.jpg2、下面这个是底物峰,产物峰。液相条件:月旭C18柱,柱温25,流动相,乙腈:磷酸盐ph8,25mmol=10:90.流速1ml/min。检测波长220nm。因为我之前对于这个方面做得很少,所以,我也不懂为什么会是这样,分不开,按照道理,应该只有3个峰出现,底物、产物、副产物http://ng1.17img.cn/bbsfiles/images/2010/04/201004182321_213084_1605610_3.jpg由于这个峰型和峰面积太难看了,我想是不是因为我的检测波长选择不对。所以第二天我就用DAD检测器来看应该选择多少波长合适。根据后面的实验结果来看,之所以分不开,不是因为流动相选择有错,而是因为产物、副产物、底物的浓度太高了。液相的响应值太高了。3、这是底物峰。流动相是:月旭C18柱,室温,21度.流动相:乙腈:25mmol磷酸缓冲盐PH3。5=10:90,DAD检测,流速,1 ml/min.http://ng1.17img.cn/bbsfiles/images/2010/04/201004182323_213085_1605610_3.jpg http://ng1.17img.cn/bbsfiles/images/2010/04/201004182324_213086_1605610_3.jpg根据3D图谱,这个底物峰在[font=Times
没用过液相色谱,也没培训过,自己在摸索。梯度洗脱怎么设置?如图,在41分钟时候溶剂从A65%B35%逐步达到A 100% B0%?还是在41分钟时候才转换成a100% b0%[img]https://ng1.17img.cn/bbsfiles/images/2021/04/202104131001220789_9197_4020217_3.png[/img]
选择R-(+)-苯乙基异氰酸酯作为手性衍生化试剂,与非索非那定生成氨基甲酸酯衍生物,通过反相高效[url=https://insevent.instrument.com.cn/t/5p][color=#3333ff]液相色谱[/color][/url]法实现对映体的分离分析。非索非那定两个对映体衍生物在25~100 ng/ml浓度范围内线性关系良好(R~2=0.9992,0.9989),日内、日间精密度均小于10%。建立的非索非那定对映体柱前手性衍生化反相高效[url=https://insevent.instrument.com.cn/t/5p][color=#3333ff]液相色谱[/color][/url]分析方法灵敏、准确,可用于体外细胞模型中盐酸非索非那定立体选择性分析。 详见姚青青等,浙江大学学报(医学版). 2014,43(02)。
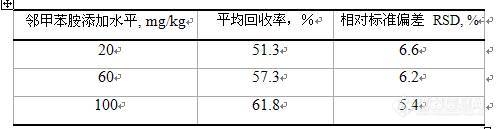
看见同行(看着这话题,绝对是同行,多交流)发着文章http://bbs.instrument.com.cn/shtml/20101012/2857370/,我这把手上的东西整整,算是补充吧。大家多多提意见吧!摘要 针对纺织品偶氮染料检验中常检出的禁用邻甲苯胺及其异构体,结合气相色谱-质谱和高效液相色谱-二极管阵列检测器(HPLC/DAD),摸索了以气相色谱-质谱进行初期定性筛选、以高效液相色谱进行确证和定量的芳香胺异构体检测方法,避免了检验中其异构体造成的假阳性检出。本方法在2 μg/mL~100 μg/mL浓度范围内线性关系良好,线性相关系数可达到0.9999;回收率为51%~62%。关键词 纺织品;检测;禁用邻甲苯胺;异构体1 前言采用气相色谱-质谱联用方法检测染料产品中24种有害芳香胺含量,按照现行国家标准中提供的气相色谱条件,会有多种异构体保留时间相同或非常接近,而且这些异构体的质谱图又非常相似,从而造成无法使用特征离子对其进行定性和定量,往往会形成假阳性结果的产生。当前针对有害芳香胺的气相色谱/质谱检测方法,大都采用非极性或极性较弱的色谱柱,如HP-5MS、DB-5MS、DB-35MS,这些色谱柱普遍存在的缺点是对常见的异构体不能很好的分离。有文献报道采用中等极性色谱柱DB - 17MS(固定相等同于50%苯甲基聚硅氧烷),对芳香胺异构体有一定的分离能力,但没能实现禁用芳香胺的基线分离,造成定量上的偏差。高效液相色谱法(HPLC)可同时测定多种芳香胺,在分离异构体上是强有力的确证方法,准确定性和定量,分析方法易于推广。本方法采用气相色谱-质谱联用对异构体进行初期定性筛选、以高效液相色谱进行确证并外标定量,对常检出的的禁用邻甲苯胺及其异构体检测进行了研究。本方法通过进一步摸索优化高效液相色谱条件,使邻甲苯胺及其异构体达到基线分离,有效避免了检测过程中的假阳性检出。2 实验材料与方法2.1 仪器和设备气相色谱/质谱联用仪:美国Agilent 公司7890A /5975 C,带自动进样器,四极杆质谱检测器;气相色谱柱:HP-5MS 5% Phenyl Methyl Silox,325 °C:30 m x 250 μ[size=
近几年,液相色谱仪器在硬件方面最大了突破就是推出了UHPLC或UPLC系统;但在色谱柱填料及形式方面也还是有一些新的技术,我收集了相关资料,简单回顾一下此方面相关的新技术。 一、色谱柱填料新技术 1、亚2微米填料 首当其冲就是很热门的亚2微米填料。根据色谱速率理论,粒径越小,柱效越高,而且当粒径小到亚2微米左右时,线速度的提高,其分离度就不再降低,而亚2微米填料的优势也正在与此。这个理论很早就有,但是为什么UPLC或UHPLC直到2004年才出现呢?原因主要是粒径减小,柱压的急剧升高,因此亚2微米填料对系统的耐压性能要求很高,长久以来,材料及工艺不能满足亚2微米填料对系统的要求。 随着材料及技术的进步,2004年沃特世推出了首款商品化地UPLC系统及配套的亚2微米填料的色谱柱。如今已有沃特世、安捷伦、赛默飞(戴安)、岛津、日立、珀金埃尔默等10余家公司推出UHPLC系统;国内上海伍丰也推出了国内首台UHPLC系统。但在填料方面,相比于常规的液相色谱填料,能够生产亚2微米色谱柱的公司还不是很多,色谱柱的种类也偏少。但毫无疑问,UPLC或UHPLC、亚2微米填料是液相色谱发展的主流。沃特世公司首席科学官Thomas E.Wheat先生认为,“未来10-15年,UPLC有可能完全取代HPLC。” 2、核壳型填料 最近,多家公司推出了一种新型液相色谱填料——核壳型填料。这种填料的优势在于,其可以缩短分析时间,提高柱效,但是对系统压力的增加却不是很多。换句话说,可以部分实现亚2微米填料的优势,但是由于对系统耐压要求不高,其可以在常规的HPLC上运行,可视为UPLC/UHPLC好的替代品。 核壳型填料就是在坚实的硅胶核心上生成一个均匀的多孔外壳。由于核心硅球是实心的,这样样品在通过色谱柱时,只需要花费少量的时间便能扩散出硅球表面的颗粒孔中,在较短时间完成扩散,更快地传质,因此分析速度及柱效都较原来普通的色谱柱有很大提高。目前,生产和供应核壳型填料的厂家有安捷伦、菲罗门、Sigma-Aldriich等。 3、整体柱(monolithic column) 整体住也是近年来液相色谱柱填料研究的又一大方向。整体柱,又称为棒状柱、连续床层、无术塞术,是一种用有机或无机聚合方法在色谱柱内进行原位聚合的连续床固定相。与常规装填的液相色谱柱相比具有更好的多孔性和渗透性,以及具有灌注色谱的特点,即色谱柱中既有流动相的流通孔又有便于溶质进行传质的中孔(几十个纳米),目前多应用于对生物大分子进行快速分离分析,应用还具有一定的局限性。目前,商品化的整体柱产品也不是很多,主要还处于科研阶段。在售的整体柱比较有名的是默克公司的ChromolithTM。 二、色谱柱新形式 1、色谱饼 色谱饼这一说法源自西北大学现代分离科学研究所、现代分离科学陕西省重点实验室耿信笃教授课题组,其与普通的色谱柱最大区别在于,其柱直径远远大于柱长,因而呈现饼的形状,故称为色谱饼。 此种形式的改变带来的好处是,色谱柱的平衡时间、进样、洗脱及色谱柱的平衡时间都显著地缩短,从而实现了快速。当然使用色谱饼的前提是,分离度不能有很大的损失。目前,这一形式的色谱柱在分析蛋白方面有很好地效果。 2、固定相优化液相色谱(Phase Optinized Liquid Chromatography POPLC) 固定相优化色谱产品来自于德国BISCHOFF公司,上海通微是该产品在中国的代理。POPLC改变了我们原来创建分析方法的传统思路,不从改变流动相或更换色谱柱来改善分析效果的角度出发,而是从“优化固定相”的角度出发来摸索分析方法。据悉,目前商品化的固定相种类非常多,如何从中选出适合的固定相是头疼的问题。研发者希望通过固定相的组合能帮助方法开发人员更快更好地找到适合的固定相。 德国BISCHOFF公司开发的POPLC系统由迷你柱管、填装不同填料的可替换短柱芯和分析软件三部分组成。在实验中,先选择几根装有不同填料的短柱芯作为一个实验组,通过单根柱子先做基础实验,然后用配套软件计算可能的柱连接方式(即将各种固定相串联),再预测分离效果,最后做验证实验。实验表明,这种方式可以有效缩短分离时间,相应提高检测灵敏度。
试验目的:为该品种后期研究如有关物质和含量测定摸索流动相。试验条件:【含量测定】照高效液相色谱法(中国药典2010年版二部附录V D)测定。色谱条件与系统适用性试验用十八烷基硅烷键合硅胶 为填充剂;取醋酸铵3.96g,加水720ml使溶解,加乙腈280ml、三乙胺10ml,用冰醋酸调节pH值至5.5为流动相;检测波长为295nm。分别取盐酸帕罗西汀、去氟帕罗两汀与N-甲基帕罗西汀对照品各5mg,置同一10ml量瓶中,加流动相 溶解并稀释至刻度,摇匀,作为系统适用性试验溶液,取20μl注人液相色谱仪,记录色谱图,出峰顺序为去氟帕罗西汀峰、盐酸帕罗西汀峰、N-甲基帕罗西汀峰,理论板数按盐酸帕罗西汀峰计算不低于3000,盐酸帕罗西汀峰、去氟帕罗西汀峰与 N-甲基帕罗西汀峰之间的分离度均应符合要求。测定法 取本品10片,精密称定,研细,精密称取适量(约相当于帕罗西汀10mg),用流动相溶解并稀释制成每 lml中约含0.lmg的溶液,摇匀,滤过,精密量取续滤液20μl注人液相色谱仪,记录色谱图;另取盐酸帕罗西汀对照品,同法测定,按外标法以峰面积计算,即得。色谱柱信息:序列号(SN):W11212195。典型色谱图:http://ng1.17img.cn/bbsfiles/images/2013/02/201302051437_424683_1621890_3.gif对照品和样品色谱峰比较:http://ng1.17img.cn/bbsfiles/images/2013/02/201302051501_424684_1621890_3.gif试验总结:采用中国药典2010年版二部收载的方法,进行原料药的含量标定,其系统适用性结果符合要求,拖尾因子和分离度不是很理想,在后期研究准备调节流动相成分比例,以期得到改善。
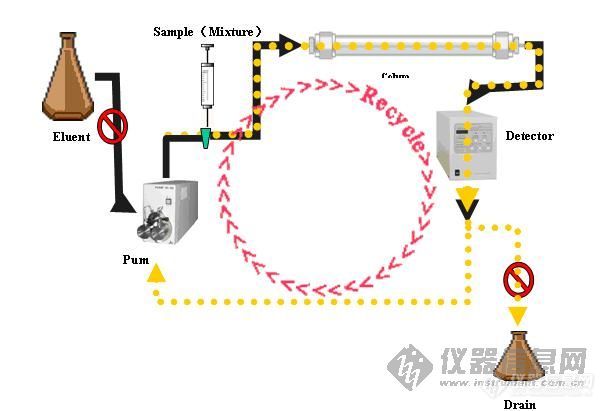
循环制备液相色谱就是较之平常的液相色谱多了个循环功能,对于一次过柱不能达到较好的分离效果的物质循环多次过柱,变相的提高了柱效,使样品达到较好的分离效果。其流程路如下:[img]http://ng1.17img.cn/bbsfiles/images/2009/08/200908140931_165538_1622597_3.jpg[/img] 由于采用了循环技术,其分离效果常规液相色谱有较大提高,对于手性物质及同分异构体也能达到很好的分离效果。对于一般样品省去了摸索最佳溶剂配比的过程,通过多次循环就能达到非常好的分离效果。利用循环功能对端基不同的样品进行分离样品为1-Benzyl-carboethoxy-4-piperidonehydrochloride (A)和1-Benzylcarbomethoxy-4-piperidonehydrochloride的混合物[img]http://ng1.17img.cn/bbsfiles/images/2009/08/200908140933_165539_1622597_3.jpg[/img]
http://simg.instrument.com.cn/bbs/images/brow/em09509.gif求助下各位大大,小弟日前用乙醇提取人面果(一种植物药材),用石油醚、正丁醇、乙酸乙酯逐步萃取,得到它们的萃取物,现在要摸索一个色谱条件把乙酸乙酯部位峰分离开,先做液相再做质谱,分析里面含啥。请教哈如何让把看看乙酸乙酯部位提取液中的峰分离开,分开收集,看大概含那些成分?请问如何分离,该选择什么样的色谱条件?谢谢了 ~~http://simg.instrument.com.cn/bbs/images/brow/emyc1004.gif
我做的是样品中的黄酮定性定量,试了许多文献的方法,只有芦丁出峰,但按理说应该还有槲皮素、山奈酚等等,一个是因为其他品种的样品有,另一个是总黄酮含量大小和芦丁含量大小不一致。在我尝试了浓缩、纯化、酸水解都不行之后,我开始摸索液相条件我看了社区的一些精华帖,选定了0-48min,5%-95%乙腈(另一个是1%乙酸),打算先测一下进样体积和流速的影响,结果发现在这个梯度下,无论怎么换别的,只有两分钟左右的溶剂峰,其余一个峰都没有。所以,我觉得可能得用分段的梯度,但是这个分段怎么分,每一段的流动相占比我不知道该怎么设置,于是就胡乱设置了一下。比如48min平分成六段,一段8分钟,让乙腈从5%开始往上升,升到100%,再走一个初始比例。我想问的是:1. 我上面的思路是对的吗?2. 大家知道梯度怎么分段设置吗?3. 有的方法混标跑的很好,槲皮素那些都能出峰,但是样品就是不出槲皮素的峰,这是不是说明我样品里就是没有这些东西?4. 我现在尝试分析别人的梯度,比如他们的槲皮素8分钟出峰,而梯度是0-10min,30-40%乙腈,这能不能说明,(40-30)/10*8+30=38,38%的乙腈可以洗脱槲皮素,我应该用38%左右的乙腈多走一会?
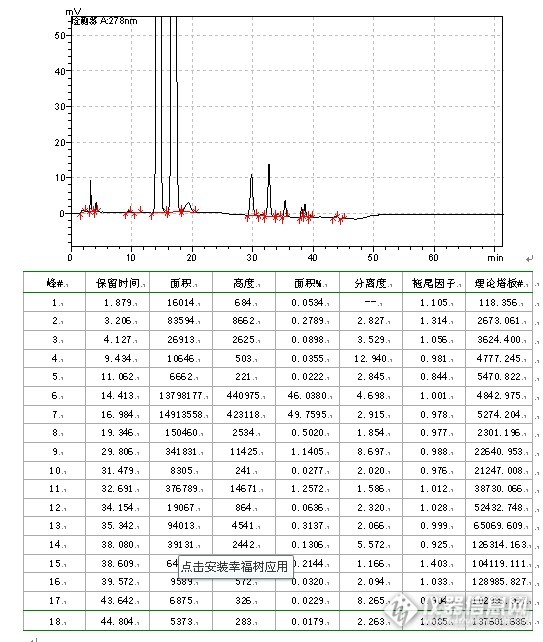
“色”路蹒跚,不拘一格,头孢呋辛酯干混悬剂有关物质流动相摸索部分。我主要工作是仿6,在遇到国家标准,我一般持研究态度,因为仿制药提倡的是仿制其质量而不是标准,一般会进行些比较。例如该品种在中国药典2010年班二部有收载,用的是等度洗脱方式,在进行研究的时候,发现梯度洗脱更具优势,所以就改变了系统方式。其具体研究方式如下:6.4 有关物质6.4.1方法的选择参照中国药典2005年版二部收载的头孢呋辛酯原料药质量标准、美国药典USP32-NF27收载的头孢呋辛酯混悬剂质量标准和新药转正标准第60册收载的头孢呋辛酯干混悬剂质量标准WS1-(X-391)-2004Z有关物质检查项,本品选择高效液相色谱法作为有关物质检测方法。6.4.2方法学验证6.4.2.1 试验材料仪器:LC-10AT、20AT VP泵(SHIMADZU CORPORATION)SPD-10A、20A、M20A VP紫外检测器(SHIMADZUCORPORATION)工作站:LC solution(SHIMADZU CORPORATION) 色谱柱:C18色谱柱,粒度5um,规格250mm×4.6mm,wel518425,LN:W1801.19,SN:1021096.主要试剂:甲醇(色谱纯)、磷酸二氢铵(分析纯)、6.4.2.2 波长选择根据专属性试验结果并参照中国药典2010年版(二部)收载的头孢呋辛酯原料药质量标准、美国药典USP32-NF27收载的头孢呋辛酯混悬剂质量标准和新药转正标准第60册收载的头孢呋辛酯干混悬剂质量标准WS1-(X-391)-2004Z有关物质检查项,本品选择高效液相色谱法作为有关物质检测方法。波长选择为278nm。6.4.2.3 流动相选择参照中国药典2010年版(二部)收载的头孢呋辛酯原料药质量标准、美国药典USP32-NF27收载的头孢呋辛酯混悬剂质量标准和新药转正标准第60册收载的头孢呋辛酯干混悬剂质量标准WS1-(X-391)-2004Z有关物质检查项色谱条件:用十八烷基硅烷键合硅胶为填充剂;流速为每分钟1.0ml,检测波长278nm,进样量20μl。6.4.2.3.1 等度洗脱流动相配制:①0.2mol/L磷酸二氢铵溶液-甲醇(60:40);②0.2mol/L磷酸二氢铵溶液-甲醇(50:50);③0.2mol/L磷酸二氢铵溶液-甲醇(55:45)。供试样品配制:称取头孢呋辛酯对照品适量(约相当于头孢呋辛11mg),置20ml量瓶中,加2ml甲醇超声溶解,分别用相应的流动相稀释至刻度,摇匀,滤过,精密量取20μl注入液相色谱仪并记录色谱图。试验结果见表10-6,色谱图见图37~39。表10-6等度洗脱试验结果 流动相异构体保留时间 (min.)拖尾因子与相邻峰 分离度理论板数出峰个数①异构体B15.7570.9922.189487111异构体A18.6880.9673.030[f
http://simg.instrument.com.cn/bbs/images/default/emyc1010.gif当你拿到一个标准后,里面的液相条件和质谱条件你还会进行摸索吗?还是只按照上面的条件设置?还有就是,前处理按照标准的方法做多少次你会判定此方法回收率做得好与坏?曾经做过一个兴奋剂的标准,质谱条件是别人摸索好的方法,前处理是自己优化出来的,大概做了十几次回收率还是50---60%,最后判断这方法如果不加内标绝对做不到像标准上说的那样1ng/ml回收率在70%---120%
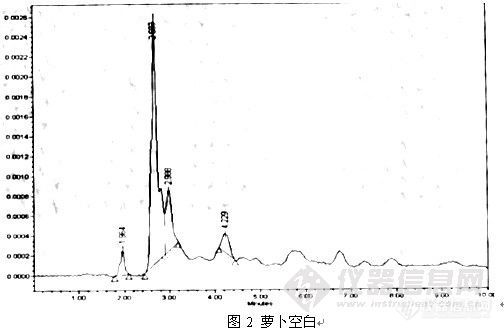
一:简介: 吡虫啉,又名脒蚜胺, 硝基亚甲基类内吸杀虫剂!主要用于防治吮吸式口器害虫!如蚜虫、飞虱、蓟马、粉虱等。关于吡虫啉的检测方法有GB/T 23379-2009 水果、蔬菜及茶叶中吡虫啉残留的测定 高效液相色谱法,SN/T 2073-2008 进出口植物性产品中吡虫啉残留最的检测方法 液相色谱串联质谱法 ,NY/T 1275-2007 蔬菜、水果中吡虫啉残留量的测定 ,SN/T 1902-2007 水果蔬菜中吡虫啉、吡虫清残留量的测定高效液相色谱法 。二:仪器条件: waters2690液相色谱仪;流动相水:乙腈65:35,流速1.0ml/min,紫外监测器270nm,进样量20微升,室温,色谱柱。ultimate xb-c18,5µm,4.6x250mmhttp://ng1.17img.cn/bbsfiles/images/2009/11/200911212331_185824_1896702_3.jpghttp://ng1.17img.cn/bbsfiles/images/2009/11/200911212331_185825_1896702_3.jpg 以前用过其他品牌的c18,可能是性质和农药不是很匹配,标样最低能做到0.1ppm,峰比较宽,增大有机相的比例,出峰就又太靠前了,3-4min出,干扰太大。用这款色谱柱按上述条件,0.1mg/kg的标样峰形较好,而且信噪比为89,标样可以做到0.02mg/kg。以前为了满足检测限,要浓缩10倍左右,现在可以浓缩2倍,甚至不浓缩,可以简化前处理过程,进一步的前处理方法正在摸索。
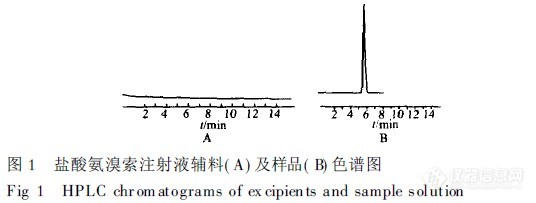
【作者】 黄淑霞; 刘金玲; 查文清; 张晓明;【机构】 深圳市南山医院; 深圳市南山医院 广东深圳518052; 广东深圳518052;【摘要】 目的:用HPLC法测定盐酸氨溴索注射液的含量。方法:采用Diamonsil-C18柱,室温下以0.04mol.L-1醋酸盐缓冲液-甲醇-乙腈(30∶40∶30)为流动相,检测波长为247nm。结果:盐酸氨溴索在5.0~100.8mg.L-1范围内呈良性线形关系,r=0.999 9,平均回收率为100.1%,RSD为0.7%。结论:该方法专属性强、简便、重现性好、结果准确可靠。 更多还原【关键词】 高效液相色谱法; 盐酸氨溴索注射液; 含量测定; http://ng1.17img.cn/bbsfiles/images/2012/08/201208131435_383503_2352694_3.jpg
http://www.instrument.com.cn/download/shtml/081950.shtmlNY/T 1379-2007 蔬菜中334种农药多残留的测定 气相色谱质谱法和液相色谱质谱法今天突然接到通知,我们市要到别的市里抽样检测神马农残和兽残,当时组长就说要用《NY/T 1379-2007 蔬菜中334种农药多残留的测定 气相色谱质谱法和液相色谱质谱法》这个方法来做,我看了一下标准,貌似标准里还没有气质的定性离子和定量离子,难到还要自己摸索?- -!偶还没试过做这些呢,上面说要做scan,但是问题是做完以后怎么选择定性离子和定量离子呢?对了,偶们的机子么有NIST05质谱库,但是是有其他的质谱库的,下午回去看看先求高手指点
跟大家分享一本书《高效液相色谱方法及应用》第二版,感兴趣的版友可以下载附件查阅,也欢迎补充。全书的目录如下:作 者: 于世林出 版 社: 化学工业出版社 本社特价书所属丛书: 色谱技术丛书 册 数: 条 形 码: 9787502569068 ; 978-7-5025-6906-8I S B N : 7502569065 出版时间: 2005-6-1开 本: 小16开 页 数: 333定 价: 39 元第一章 绪论第一节 高效液相色谱法的特点一、与经典液相(柱)色谱法比较二、与气相色谱法比较三、高效液相色谱法的优点四、高效液相色谱方法发展简介第二节 高效液相色谱法的分类一、按溶质在两相分离过程的物理化学原理分类二、按溶质在色谱柱洗脱的动力学过程分类第三节 高效液相色谱法的应用范围和局限性一、应用范围二、方法的局限性参考文献第二章 高效液相色谱仪简介第一节 流动相及储液罐一、储液罐二、流动相脱气第二节 高压输液泵及梯度洗脱装置一、高压输液泵二、输液系统的辅助设备三、梯度洗脱装置第三节 进样装置一、停流进样装置二、六通阀进样装置三、自动进样器第四节 色谱柱一、柱材料及规格二、柱填料三、保护柱四、柱连接方式五、柱温控制第五节 检测器一、检测器的分类和响应特性二、紫外吸收检测器三、折光指数检测器四、电导检测器五、荧光检测器六、蒸发光散射检测器第六节 色谱数据处理装置一、微处理机二、色谱工作站参考文献第三章 液固色谱法和液液色谱法第一节 分离原理一、吸附系数二、分配系数第二节 固定相一、液固色谱固定相二、液液色谱固定相第三节 流动相一、表征溶剂特性的重要参数二、液固和液液色谱的流动相第四节 二元溶剂体系中液固和液液色谱的保留规律一、溶质保留值的基本方程式二、液固色谱的保留值方程式三、液液色谱的保留值方程式参考文献第四章 键合相色谱法第一节 分离原理一、正相键合相色谱法的分离原理二、反相键合相色谱法的分离原理第二节 固定相一、键合固定相的制备及分类二、键合固定相的性质三、使用键合固定相应注意的问题第三节 流动相一、溶剂的选择性分组二、在键合相色谱中选择流动相的一般原则三、改善色谱分离选择性的方法四、多元混合溶剂的多重选择性五、溶质保留值随溶剂极性变化的一般保留规律六、用线性溶剂化自由能关系(LSER)来表征反相液相色谱中溶质的保留值方程式第四节 新型高效液相色谱的固定相和流动相一、新型高效化学键合固定相二、化学键合固定相分类方法简介三、整体色谱柱四、超热水流动相第五节 离子对色谱法一、分离原理二、固定相、流动相和对(反)离子三、影响离子对色谱分离选择性的因素参考文献第五章 梯度洗脱第一节 基本原理一、等度洗脱二、梯度洗脱第二节 影响梯度洗脱的各种因素一、梯度洗脱时间(tG)对分离的影响二、强洗脱溶剂组分B浓度变化范围的影响三、梯度陡度对保留值的影响四、柱温变化对保留值的影响五、梯度洗脱程序曲线形状的影响六、影响梯度洗脱的其他变量第三节 优化梯度洗脱的方法一、建立梯度洗脱方法的一般步骤二、梯度洗脱中的实验条件第四节 梯度洗脱的图示方法一、二元溶剂梯度洗脱二、三元溶剂梯度洗脱三、四元溶剂梯度洗脱四、用极坐标和球面坐标描述梯度洗脱参考文献第六章 体积排阻色谱法第一节 分离原理一、分布系数二、体积排阻色谱法的特点第二节 固定相一、固定相的分类二、凝胶固定相的特性参数三、凝胶色谱柱的制备及谱图特点第三节 流动相一、凝胶渗透色谱的流动相二、凝胶过滤色谱的流动相第四节 凝胶渗透色谱法测定聚合物分子量分布一、聚合物分子量、分子量分布及测定的意义二、凝胶渗透色谱图的解析及数据处理参考文献第七章 高效液相色谱法的基本理论第一节 表征液相色谱柱填充性能的重要参数一、总孔率二、柱压力降三、柱渗透率第二节 高效液相色谱的速率理论一、影响色谱峰形扩展的各种因素二、范第姆特方程式的表达及图示第三节 诺克斯方程式一、描述色谱柱性能的折合参数二、诺克斯方程式第四节 色谱柱操作参数的优化一、三个柱操作参数的表达式二、HPLC中实用柱操作参数的优化三、柱操作参数优化的图示表达方法第五节 “无限直径”效应和柱外效应一、“无限直径”效应二、柱外效应第六节 超高效液相色谱一、超高效液相色谱的理论基础二、实现超高效液相色谱的必要条件三、超高效液相色谱的应用参考文献第八章 高效液相色谱分离条件的优化第一节 高效液相色谱中色谱参数的相关性一、色谱参数的分类二、色谱参数的相关性第二节 色谱分离条件优化标准的选择一、难分离物质对的峰对分离优化标准二、整体色谱图的优化标准第三节 色谱响应函数和色谱优化函数一、Morgan和Deming提出的色谱响应函数二、Watson和Carr提出的色谱响应函数三、Glajch和Kirkland提出的色谱优化函数四、Berridge提出的色谱响应函数第四节 色谱分离条件的优化方法一、单纯形法二、窗图法三、混合液设计实验法四、重叠分离度图法五、等强度洗脱和梯度洗脱的优化图示法第五节 优化HPLC分离的计算机辅助方法一、实验设计系统二、人工智能系统第六节 高效液相色谱专家系统简介一、专家系统的组成二、专家系统的使用方法参考文献第九章 微柱液相色谱法第一节 方法简介一、微型柱的分类二、微柱液相色谱法的优点和缺点第二节 基本理论一、柱外效应二、管壁效应三、稀释效应四、分离阻抗第三节 仪器装置一、输液泵系统二、进样系统三、柱系统四、检测器系统五、连接管和接头第四节 微柱的制备一、评价微柱性能的重要参数二、影响微柱分离效率的相关参数三、微柱的制备方法第五节 微柱液相色谱的新技术一、纳米液相色谱技术二、超高压液相色谱技术参考文献第十章 二维高效液相色谱法第一节 描述分离体系效能的参数一、峰容量二、信息量第二节 二维高效液相色谱的技术功能一、切割功能二、反冲洗脱功能三、痕量组分的富集功能第三节 二维高效液相色谱的流路系统一、多通路切换阀二、二维高效液相色谱的流路系统第四节 二维高效液相色谱在蛋白质组学研究中的应用参考文献第十一章 建立高效液相色谱分析方法的一般步骤和实验技术第一节 样品的性质及柱分离模式的选择一、样品的溶解度二、样品的分子量范围三、样品的分子结构和分析特性第二节 分离操作条件的选择一、容量因子和死时间的测量二、色谱柱操作参数的选择三、样品组分保留值和容量因子的选择四、相邻组分的选择性系数和分离度的选择第三节 高效液相色谱法的实验技术一、溶剂的纯化技术二、色谱柱的装填技术三、色谱柱的平衡、保护与清洗、再生技术四、梯度洗脱技术五、色谱柱前和柱后的衍生化技术六、样品的预处理技术参考文献符号表
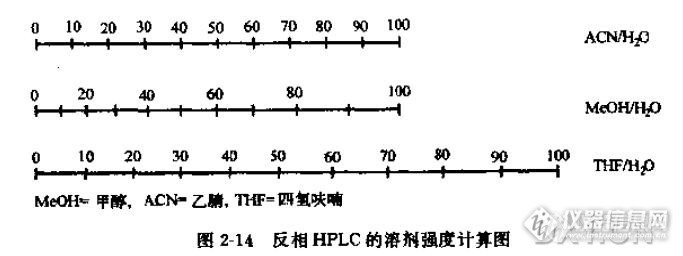
在沉寂了2年之后,qingqingcao再次探讨高效液相色谱。这次和大家探讨一些关于HPLC方法学开发的问题。当然,这是很久以前的思路,在今天看来是比较幼稚的,但是,如果能够给大家一点哪怕一点点启示也是好的。当然文章有很多缺陷,也希望大家批评。高效液相色谱在极性的角度可以分成正相色谱分析和反相色谱分析。反相色谱分析一般使用水-甲醇-乙腈体系,一般选用C8,C10,C18 柱子,主要使用C18键合相柱子。非极性物质一般用氯仿-正己烷体系。因为qingqingcao没有做过正相HPLC分离,所以,我们主要关注反相HPLC分析。色谱柱的选择当然,选用的色谱柱一般就是C18柱。长度呢?如果分析的物质复杂,那么可以选取长一些的,比如250mm╳4.6mm╳5μm。保证其分离度。 如果物质不是很复杂,那么选取150mm╳4.6mm╳5μm的色谱柱,可以有效地缩短分析时间。检测器的选择紫外检测器是最通用的检测器之一,所以,本文均以紫外检测器做说明。对于需要分析的物质,做全波长扫描,由于物质的不同官能团,他们会在不同的地方有吸收。通过综合选取大家都有吸收的波长。选择波长需要有权衡。同时也可以用一些特殊的波长,避免杂质的吸收,也可以提升测试准确度。或者使用双波长的方法。流动相的选择对于选取流动相,首先实验一下样品的溶解度。样品在水中,甲醇,乙腈中溶解程度。如果,很容易溶解在水中,这就告诉我们,流动相选取,可能有机相要少些,比方说测试维生素C,其极性很强,流动相中有机相比例不能多。(为了保护色谱柱,有机相比例一般不少于8% )如果,样品不太容易溶解在水中,那么有机相的比例应高些,比方像维生素E,就可以用90% -95% 的甲醇体系。其次是样品的酸碱度。我们知道,C18色谱柱流动相pH在2-8 之间。太酸,太碱都可能损坏色谱柱。流动相pH值对于分离有一定作用,所以,酸性物质,一般流动相酸一些,碱性物质可能碱一些。对于几种物质的分离。如果不是太多的物质,用等度方法会比较好。流动相一般开始选用20mM磷酸缓冲盐-乙腈体系。(pH一定,流速一定)。因为乙腈洗脱好,而且粘度低。通过水相-有机相的比例的改变,观察各色谱峰的分离情况。这样可以运用无限夹逼法找到最合适的值。比方说,35:65(ACN:磷酸盐)达到分离,但是还是有部分叠加,那么就微调即可。当然还要考虑到分析时间问题。不要为了完全分离而牺牲了分析时间。所以,在色谱分析,有一个k值(容量因子)。κ∈(2,20)如果真的无法改变,那么可以试着微调pH值,来分离。当然,由于乙腈毒性强,所以通过一定计算,用甲醇同等地接替乙腈。http://ng1.17img.cn/bbsfiles/images/2015/09/201509251447_567900_1626663_3.jpg如果有些物质很靠近死体积,那么需要考虑添加0.02mM离子对试剂,加强物质的保留能力。流速一般液相流速在0.8-1.4ml/min。首选1.0ml/min柱温柱温不影响色谱的分离,但是,相对稳定的温度,可以使得保留时间稳定。进样量手动进样,只有进样环。一般也就是20μL。自动进样的选择很多。但是,最好不要超过80μL。因为进样多,容易造成展宽。分离物质的浓度:分离物质,不能够太浓也不能太稀。太浓,容易过载;太稀检测不出。具体还是根据检测器的响应来调整。总结一下HPLC分析,首先要找到合适的流动相。流动相既要保证分离,又要关心分析时间。流动相尽量使用毒性小一点的甲醇。根据样品和标准品的PH值进行调节流动相溶液酸碱度。检测波长很重要,要懂得合适的波长。也就是说,这个单一波长可以分析出所有需要分析的物质。可以通过波长的调整,规避杂质峰的干扰。温度和流速都对分离,影响不大。分离条件摸索好后,就可以开展1)线性浓度测试2)精密度测试3)样品测试4)回收率测试5)空白测试。6)最小检测限,最小检测量定量的工作比较简单,就是按照以上的思路去操作。那么一个项目就这么完成了。当然,这些需要我们的工程师们的努力和智慧。也许,qingqingcao也不再写色谱方面的文章了;因为我没有机会去碰HPLC仪器了。那么,就拿这篇文章作为一个纪念吧。想想从2002年春天,还是一个大学生,去做HPLC,笨手笨脚的,导师也经常批评我。后来又接触了不少的HPLC仪器,也做过维修维护,开发过方法,做过常规工作……这么多年了,一个生手,变成一个熟练工,也能做一点点方法开发,青春就贡献在HPLC上了。看着论坛上有许多HPLC高手,他们的文章,让我觉得,我们时代的发展。后浪推着前浪。后浪必定会超过前浪的。我也希望,我在论坛上写的这些东西,能够给初学者一点点启示,当然,如果有错误的话,希望不会误导大家。如果能够记住qingqingcao做了一点事情,曾经在HPLC上做过点事情,我也欣慰了。也谢谢仪器信息网官人们的努力工作,为我们搭起这样一个平台。
[color=#444444]方法采用的是孙宝利等的《高效液相色谱法测定土壤中有机酸》(2010),具体:A浓度标准品为草酸:苹果酸:柠檬酸:乙酸=0.2:5:5:5(草酸0.2ug/ml,其它3种5ug/ml),流动相:0.1%磷酸:乙腈=98:2,流速1mL/min,波长210nm,进样量20μL,柱温35℃。测的时候混标的峰测出来不明显,应该说是找不到各标样的峰,于是我做了单标,但是单标的峰还是找不到,有少许小的杂峰,而且每个标样和样品在进样的第12min时都有一个固定的峰值,不知道是怎么回事。 [/color][color=#444444]另外文献中说的流动相的ph为2.2,而我当时没有调到2.2,所以我准备将ph重新调一下,请问是直接加盐酸将0.1%磷酸溶液的ph调到2.2吗?[/color][color=#444444]我们所也没有人测过有机酸,所以目前我还在摸索中,但遇到这些问题我也不知是什么原因[/color]
本法用甲醇-水为流动相,在C18反相柱上建立了陆英中具有乌索酸含量测定的高效液相色谱法,以95%乙醇为溶剂处理样品具有提取率高、色谱干扰小、物质分离效果好的优点,本法简便易行、快速准确,其最小检出限为0.2ug。不同浓度水平检测结果日间、日内相对误差小于4.0%。关键词:陆英草 乌索酸 高效液相色谱 含量测定Analysis of Ursolic Acid in Herba Sambuci Chinensis using HplcThe paper reported the determination of Ursolic Acid inHerba Sambuci Chinensis using Hplc,the separation was achieved by applying an Waters -ODS 150×4.6 mm (5um) column and methanol-water (90:10) as mobile phase at flow rate of 0.8 ml/min. the UV detector was set at 210nm External standard method was applied .The linear range was 150-2000ug/ml with the lower limit of detection of 0.2ug.It was found to be effielent and low interference to extract the samples with ether.陆英(Herba Sambuci Chinensis )系为忍冬科接骨木属植物,生于山坡、路旁、溪边、荒野灌丛中。产于长江以南地区。 据报道[1]陆英草含氯原酸、α-香树脂素棕榈酸酯(α-amyin palmitate)、乌索酸、β-谷甾醇、豆甾醇、油菜甾醇、硝酸钾、黄酮、鞣质等。目前其它中药材乌索酸含量的测定多采用比色法及薄层扫描[2]以及衍生化法[url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]检测[3],其操作繁琐,显色及前处理误差大,费事费力。本文采用高效液相色谱法用甲醇-水为流动相,检测波长210nm,在C18反相色谱柱上建立了其含量的测定方法,具有简便可行、准确、快速、物质分离效果好的优点,重现性好,适应范围广,利于对陆英草的准确、深入研究。实验部分一、 仪器设备及试剂Waters 600E Hplc系统,UV-2487 可变波长检测器 ,20ul定量环,M32数据处理工作站。乌索酸对照品:中国医学科学院药物研究所 95%乙醇(AR) 甲醇(AR) 水二、实验方法1、色谱条件 色谱柱:Waters -ODS 150×4.6 mm (5um);流动相:甲醇-水 (90:10); 流速:0.8 ml/minUV波长:210nm ;量程:0.005 AUFS ;温度:25℃数据处理:峰面积外标定量。2、样品处理 (1)将原料(樟树医药公司)的叶粉碎过筛并恒重。(2)称取样品10克,置索氏提取器中,加95%乙醇回流提取8小时,提取液浓缩定容于100毫升的容量瓶中,取上清液约5毫升过针式过滤器过滤,取滤液待测。3、标准曲线的确定分别吸取配好的5.0mg/ml乌索酸标准液0.4、0.8、1.6、2.8、3.6ml置于10ml容量瓶中,用甲醇稀释成 0.2、0.4、0.8、1.4、1.8mg/ml系列标准溶液,在选定的色谱条件下,重复进3次每次20ul,以标准品峰面积与浓度关系得出回归方程:Y=3.35 E+005X+8.74E+003 R=0.99994、样品测定 将预处理好的待测样品液进样20 ul 检测,通过软件操作的得出浓度值。三、结果1、乌索酸对照品与陆英样品的色谱图1 图1 图22、方法重现性 用两个浓度水平进行连续和间隔时间及连续3天测定,考察方法重现性。结果:其最大相对标准偏差小于4.0%。2、加标回收率 吸取5.0mg/ml乌索酸标准液 0.8、1.6、3.6各三份,分别加入原料样品10克,按前法操作,测定结果,计算回收率结果为99.9%-100.8%,平均100.4%,RSD为1.0%。讨论1、迷迭香中的乌索酸是非极性五环三萜类酸,在C18反相柱上有较大的保留值,以甲醇和水二元体系作为流动相,甲醇浓度与极性相近共存物质的分离及峰形有显著影响,本法选定比例是考虑实际样品组分分离确定的,对于其它品种样品可作适当的调整。2、检测波长 我们进行了波长扫描,乌索酸在205nm处有最大吸收峰,本法采用210nm,恒流比流动相洗脱对检测无影响。3、本法检测出陆英草中平均含0.28%的乌索酸。4、本法简便、易行,准确快速。







 我要推广仪器
我要推广仪器
 下载APP
下载APP