国标中空白试验的定义空白试验:除不加试样外,采用完全相同的分析步骤、试剂和用量(滴定法中标准滴定液的用量除外),进行平行操作所得的结果。用于扣除试样中试剂本底和计算方法的检出限。空白做不好会怎样?空白试验测得的结果称为空白试验值。在进行样品分析时所得的值减去空白试验值得到的才是最终分析结果。空白试验是实验室质量控制的重要环节,其准确性对提高检测结果的准确度至关重要。空白试验值反应了测试仪器的噪声、试剂中的杂质、环境及操作过程中的沾污等因素对样品测定产生的综合影响,直接关系到测定的最终结果的准确性。空白试验值低,数据离散程度小,分析结果的精度随之提高,它表明分析方法和分析操作者的测试水平较高。当空白试验值偏高时,应全面检查试验用水、试剂、量器和容器的沾污情况、测量仪器的性能及试验环境的状态等,以便尽可能地降低空白试验值。做酸碱滴定时为什么要做空白试验?因为用作稀释液的空白溶液有一定的酸碱度,会影响滴定结果,所以要做空白试验,来扣除空白的干扰。比如空白溶液为酸性,这时候用碱滴定此溶液,得到的结果会偏高,要扣除空白溶液的酸度,得到的酸度才正确。[b][color=#ac1d10]石墨炉测铅时,空白[color=#888888](4硝酸+1高氯酸GR)[/color]值总是较高,与灰化法的结果不大一致(样品为植物样)。[/color][/b][color=#595959]【分析】1. 所用的水为用石英亚沸水蒸馏器蒸馏得到的,先打一下空白,一般不会超过0.0015,然后采用的为硝酸(工艺超纯)和高氯酸(优极纯)消化,最后溶解用的1摩尔每升的盐酸或硝酸(结果差不多,只是盐酸稳定性要好一点),定容体积为50mL的话空白值一般为0.03左右。不过铅比较难做,基体干扰很大。[/color][color=#595959]2. 空白问题来自多方面,上面说的水与试剂外,你用的氩气是高纯的吗?也可用高纯氮气,但要注意分子带背景[/color][color=#595959]3. 可能来自由你所用的硝酸和高氯酸不纯所致。你可以先测空管,然后测你所用的水,再测含酸的水空白看看就知道了[/color][color=#595959]4.有可能是试液的酸度过大会影响测定,特别是使用高氯酸,影响更大,酸度大对石墨炉损害也比较大。对于石墨炉测定铅,湿法消化最好使用微波消化,使用硝酸和双氧水,这样空白中酸度比较容易控制,空白也比较低。[/color][color=#595959]5. a.实际Pb含量有出入,厂家就没有测准;b.你的仪器可能没有调制最佳。[/color]原子荧光空白偏高原因1.测定介质的选择及浓度的影响选择HNO3、HCl、HClO4、H3PO4和H2SO4等进行了实验,结果表明:以HClO4和H3PO4作介质响应值较低,汞在H2SO4溶液中能得到较大灵敏度,但空白值也高。汞在HNO3和HCl溶液中的荧光强度差别不大。在5%一25%的硝酸和5%一20%的盐酸中荧光强度基本稳定。[b][/b]2.酸的影响作为原子荧光的载流和标准试剂的基体,酸对原子荧光的影响十分的突出。一般在原子荧光中使用的酸主要有两种-盐酸和硝酸。我们习惯上使用盐酸。如果购买市场上质量差的酸,对空白的影响要大得多。[b][/b]3.还原剂的影响作为原子荧光的还原剂,一般使用最多的是硼氢化钾或硼氢化钠,在原子荧光法中,还原剂对样品以及空白值的影响主要体现在它的用量。硼氢化钾浓度不宜超过3.0%。实验表明,当硼氢化钠浓度为1%时,灵敏度和稳定性好,并且有效消除干扰。[b][/b]4.灯电流的影响一般来说灯电流与负高压对原子荧光的影响是同方向的。提高灯电流,空白值就相应的提高。空白值太大的话,可以适当的降低电流值,使仪器的灵敏度降低,减少标准空白的背景值,使所测的数据准确、可靠,但是如果电流过小,灵敏度也将变小,对所测样品的影响就会增加,所以选择合适的灯电流,保证一定的空白值与灵敏度才是关键。[b][/b]5.载气的影响实验表明,当氩气流速较低时,测定的灵敏较高.但结果的变动性加大,实际测定取氩气流速200ml/hifn,此时测定的灵敏度较高,相对标准偏差可以接受。空白色谱柱出峰,什么原因?[align=center][/align][i][color=#595959]高效液相色谱,色谱柱是Kromasil C18(15cm*5Um),流动相为10%乙腈和90%三氟乙酸水,走空白色谱柱时在整个保留时间的中间位置出现较大的色谱峰,走了好几针空白,每针的响应都不同,有的很高,有的又很低,但杂峰一直存在,不像是色谱柱里有杂质,用流动相冲洗后依然出现此问题,什么原因?[/color][/i][b][color=#595959][/color][color=#3e3e3e]【原因】[/color][/b][color=#3e3e3e]1、确定检测波长,如果是末端吸收,排除三氟乙酸的干扰。[/color][color=#3e3e3e]2、使用100%乙腈作为流动相,乙腈作为样品进样,如果为基线,则说明色谱柱及系统完好。[/color][color=#3e3e3e]3、然后在换流动相为10%乙腈和90%三氟乙酸水,样品为乙腈,如果有上述“中间位置出现较大的色谱峰”,那说明八成问题在三氟乙酸上,或者稀释三氟乙酸的水上。[/color][color=#3e3e3e]4、如果上述2、3都有“中间位置出现较大的色谱峰”,检查检测器及进样器。[/color][color=#3e3e3e]5、这些还不能解决,请联系工程师[/color][color=#3e3e3e][/color]空白乙腈出峰,什么原因?[i][color=#595959]岛津高效液相色谱仪,进空白乙腈,在样品峰位子出现倒峰,进双蒸水在样品峰出现正峰,进样品时有杂峰干扰,我已经排除了水和乙腈的问题,不知道是不是进样池或者检测器污染了,已经整了10多天,还是没效果,请指教原因和解决办法。[/color][/i][b]【分析】[/b]液相中出现倒峰,主要原因是样品吸收比流动相吸收小,所以排除影响,不仅要考虑进样系统污染和检测池污染,还要考虑流动相和色谱柱的影响。换一根柱子试试,考察系统的情况,如果照旧,就排除色谱柱的影响,然后更换流动相试试,再以后清洗进样系统和检测池等等。或将色谱柱从系统中断开,直接用乙腈或水做流动相清洗检测器就可以知道是否是检测器受污染,如果基线正常,再检查你的进样系统,样品,前处理是否有问题,如果都没问题,就去检查色谱柱吧。[color=#ac1d10][b]水质分析中氨氮的测定空白值很高的原因[/b][/color][align=center][/align]实验中纳氏试剂和酒石酸钾钠对空白值的高低影响很大,可能原因如下:1.测定使用的蒸馏水被污染,含有影响实验结果的杂质,比如蒸馏水含氨。2.试管、移液管等仪器没有清洗彻底,这些因素对实验结果也有较大的影响。3.实验所用的无氨水质量不佳,含氨量比较高,导致实验得到的空白值偏高,这是最主要的原因。4.在配制酒石酸钾钠和钠氏试剂时出现较大偏差,导致实验所用的试剂纯度(酒石酸钾钠、钠氏试剂)不佳,从而导致实验得到的空白值偏高。总氮空白值偏高,什么原因?1、试剂的配制碱性过硫酸钾的配制过程十分重要,掌握不好,会影响消解效果,对测定结果产生一定的影响。配制该溶液时,可分别称取过硫酸钾和氢氧化钠,两者分开配制,再混合定容,或者先配制氢氧化钠溶液,待其温度降到室温后再加入过硫酸钾溶解。若二者在一只烧杯中溶于水,应缓慢加水,同时搅拌,防止氢氧化钠放热使溶液温度过高引起局部过硫酸钾失效。2、玻璃器皿的洗涤[b][/b]所使用的玻璃器皿应先用(1+9)盐酸浸泡后,再用无氨水冲洗数次才能使用,否则,也会造成空白值偏高或平行性较差的情况。3、比色时的注意事项该项目的测定涉及两个波长(220nm和275nm),建议在测定完一组样品的同一波长后,再调整到另一波长,统一测定,不要测完一个样品的两个吸光度后再换另一个样品,这样反复调整波长会引起一定的测量误差。4、试剂的选择碱性过硫酸钾消解紫外分光光度法测定总氮的过程中,过硫酸钾是至关重要的试剂。首先,试剂的纯度关系到空白值的高低、测定结果的准确度。一般普通分析纯过硫酸钾的总氮含量最高不超过0.005%,但由于试剂质量存在差异,有些厂家、批次的试剂含氮量常常达不到这个要求,致使空白值偏高。因此,有条件的话建议使用优级纯试剂,尽量降低试剂中的含氮量,从而降低实验空白值。5、实验用水及试剂的质量检验[b][/b]若实验的空白值不够理想,则需要对实验用水及试剂进行检验,以选择出含氮量最低的水和试剂,获得理想的空白值。[color=#ac1d10][b]粗蛋白测定空白偏高的影响因素?[/b][/color]可以从以下方面考虑:1.配溶液的水换没换;2.看看消化炉的回收率;3.看看蒸馏仪的回收率;4.盐酸在不在保质期内;5.配溶液的硫酸是不是有问题;6.检查催化剂,是不是含有硝酸钾。小结空白试验是实验室质量控制的重要环节,其准确性对提高检测结果的准确度至关重要。以分光光度法空白为例 ,要用相应的实验用纯水或有机溶济作参比,不能用空白实验溶液做参比,以免人为减小空白值,从而掩盖空白实验值的变异 。值得注意的是,通过计量认证的实验室,由于检测仪器和玻璃量器定期检定 ,以及采取了全面的质量管理措施,因此,若测定的空白实验值太大,一般是化学药品纯度不够,配制的试液变质或被污染等原因 。应重新配制试液 ,返工重测该批样品 ,直至空白实验值合格。国标中空白试验的定义空白试验:除不加试样外,采用完全相同的分析步骤、试剂和用量(滴定法中标准滴定液的用量除外),进行平行操作所得的结果。用于扣除试样中试剂本底和计算方法的检出限。空白做不好会怎样?空白试验测得的结果称为空白试验值。在进行样品分析时所得的值减去空白试验值得到的才是最终分析结果。空白试验是实验室质量控制的重要环节,其准确性对提高检测结果的准确度至关重要。空白试验值反应了测试仪器的噪声、试剂中的杂质、环境及操作过程中的沾污等因素对样品测定产生的综合影响,直接关系到测定的最终结果的准确性。空白试验值低,数据离散程度小,分析结果的精度随之提高,它表明分析方法和分析操作者的测试水平较高。当空白试验值偏高时,应全面检查试验用水、试剂、量器和容器的沾污情况、测量仪器的性能及试验环境的状态等,以便尽可能地降低空白试验值。做酸碱滴定时为什么要做空白试验?因为用作稀释液的空白溶液有一定的酸碱度,会影响滴定结果,所以要做空白试验,来扣除空白的干扰。比如空白溶液为酸性,这时候用碱滴定此溶液,得到的结果会偏高,要扣除空白溶液的酸度,得到的酸度才正确。[b][color=#ac1d10]石墨炉测铅时,空白[color=#888888](4硝酸+1高氯酸GR)[/color]值总是较高,与灰化法的结果不大一致(样品为植物样)。[/color][/b][color=#595959]【分析】1. 所用的水为用石英亚沸水蒸馏器蒸馏得到的,先打一下空白,一般不会超过0.0015,然后采用的为硝酸(工艺超纯)和高氯酸(优极纯)消化,最后溶解用的1摩尔每升的盐酸或硝酸(结果差不多,只是盐酸稳定性要好一点),定容体积为50mL的话空白值一般为0.03左右。不过铅比较难做,基体干扰很大。[/color][color=#595959]2. 空白问题来自多方面,上面说的水与试剂外,你用的氩气是高纯的吗?也可用高纯氮气,但要注意分子带背景[/color][color=#595959]3. 可能来自由你所用的硝酸和高氯酸不纯所致。你可以先测空管,然后测你所用的水,再测含酸的水空白看看就知道了[/color][color=#595959]4.有可能是试液的酸度过大会影响测定,特别是使用高氯酸,影响更大,酸度大对石墨炉损害也比较大。对于石墨炉测定铅,湿法消化最好使用微波消化,使用硝酸和双氧水,这样空白中酸度比较容易控制,空白也比较低。[/color][color=#595959]5. a.实际Pb含量有出入,厂家就没有测准;b.你的仪器可能没有调制最佳。[/color]原子荧光空白偏高原因1.测定介质的选择及浓度的影响选择HNO3、HCl、HClO4、H3PO4和H2SO4等进行了实验,结果表明:以HClO4和H3PO4作介质响应值较低,汞在H2SO4溶液中能得到较大灵敏度,但空白值也高。汞在HNO3和HCl溶液中的荧光强度差别不大。在5%一25%的硝酸和5%一20%的盐酸中荧光强度基本稳定。[b][/b]2.酸的影响作为原子荧光的载流和标准试剂的基体,酸对原子荧光的影响十分的突出。一般在原子荧光中使用的酸主要有两种-盐酸和硝酸。我们习惯上使用盐酸。如果购买市场上质量差的酸,对空白的影响要大得多。[b][/b]3.还原剂的影响作为原子荧光的还原剂,一般使用最多的是硼氢化钾或硼氢化钠,在原子荧光法中,还原剂对样品以及空白值的影响主要体现在它的用量。硼氢化钾浓度不宜超过3.0%。实验表明,当硼氢化钠浓度为1%时,灵敏度和稳定性好,并且有效消除干扰。[b][/b]4.灯电流的影响一般来说灯电流与负高压对原子荧光的影响是同方向的。提高灯电流,空白值就相应的提高。空白值太大的话,可以适当的降低电流值,使仪器的灵敏度降低,减少标准空白的背景值,使所测的数据准确、可靠,但是如果电流过小,灵敏度也将变小,对所测样品的影响就会增加,所以选择合适的灯电流,保证一定的空白值与灵敏度才是关键。[b][/b]5.载气的影响实验表明,当氩气流速较低时,测定的灵敏较高.但结果的变动性加大,实际测定取氩气流速200ml/hifn,此时测定的灵敏度较高,相对标准偏差可以接受。空白色谱柱出峰,什么原因?[align=center][/align][i][color=#595959]高效液相色谱,色谱柱是Kromasil C18(15cm*5Um),流动相为10%乙腈和90%三氟乙酸水,走空白色谱柱时在整个保留时间的中间位置出现较大的色谱峰,走了好几针空白,每针的响应都不同,有的很高,有的又很低,但杂峰一直存在,不像是色谱柱里有杂质,用流动相冲洗后依然出现此问题,什么原因?[/color][/i][b][color=#595959][/color][color=#3e3e3e]【原因】[/color][/b][color=#3e3e3e]1、确定检测波长,如果是末端吸收,排除三氟乙酸的干扰。[/color][color=#3e3e3e]2、使用100%乙腈作为流动相,乙腈作为样品进样,如果为基线,则说明色谱柱及系统完好。[/color][color=#3e3e3e]3、然后在换流动相为10%乙腈和90%三氟乙酸水,样品为乙腈,如果有上述“中间位置出现较大的色谱峰”,那说明八成问题在三氟乙酸上,或者稀释三氟乙酸的水上。[/color][color=#3e3e3e]4、如果上述2、3都有“中间位置出现较大的色谱峰”,检查检测器及进样器。[/color][color=#3e3e3e]5、这些还不能解决,请联系工程师[/color][color=#3e3e3e][/color]空白乙腈出峰,什么原因?[i][color=#595959]岛津高效液相色谱仪,进空白乙腈,在样品峰位子出现倒峰,进双蒸水在样品峰出现正峰,进样品时有杂峰干扰,我已经排除了水和乙腈的问题,不知道是不是进样池或者检测器污染了,已经整了10多天,还是没效果,请指教原因和解决办法。[/color][/i][b]【分析】[/b]液相中出现倒峰,主要原因是样品吸收比流动相吸收小,所以排除影响,不仅要考虑进样系统污染和检测池污染,还要考虑流动相和色谱柱的影响。换一根柱子试试,考察系统的情况,如果照旧,就排除色谱柱的影响,然后更换流动相试试,再以后清洗进样系统和检测池等等。或将色谱柱从系统中断开,直接用乙腈或水做流动相清洗检测器就可以知道是否是检测器受污染,如果基线正常,再检查你的进样系统,样品,前处理是否有问题,如果都没问题,就去检查色谱柱吧。[color=#ac1d10][b]水质分析中氨氮的测定空白值很高的原因[/b][/color][align=center][/align]实验中纳氏试剂和酒石酸钾钠对空白值的高低影响很大,可能原因如下:1.测定使用的蒸馏水被污染,含有影响实验结果的杂质,比如蒸馏水含氨。2.试管、移液管等仪器没有清洗彻底,这些因素对实验结果也有较大的影响。3.实验所用的无氨水质量不佳,含氨量比较高,导致实验得到的空白值偏高,这是最主要的原因。4.在配制酒石酸钾钠和钠氏试剂时出现较大偏差,导致实验所用的试剂纯度(酒石酸钾钠、钠氏试剂)不佳,从而导致实验得到的空白值偏高。总氮空白值偏高,什么原因?1、试剂的配制碱性过硫酸钾的配制过程十分重要,掌握不好,会影响消解效果,对测定结果产生一定的影响。配制该溶液时,可分别称取过硫酸钾和氢氧化钠,两者分开配制,再混合定容,或者先配制氢氧化钠溶液,待其温度降到室温后再加入过硫酸钾溶解。若二者在一只烧杯中溶于水,应缓慢加水,同时搅拌,防止氢氧化钠放热使溶液温度过高引起局部过硫酸钾失效。2、玻璃器皿的洗涤[b][/b]所使用的玻璃器皿应先用(1+9)盐酸浸泡后,再用无氨水冲洗数次才能使用,否则,也会造成空白值偏高或平行性较差的情况。3、比色时的注意事项该项目的测定涉及两个波长(220nm和275nm),建议在测定完一组样品的同一波长后,再调整到另一波长,统一测定,不要测完一个样品的两个吸光度后再换另一个样品,这样反复调整波长会引起一定的测量误差。4、试剂的选择碱性过硫酸钾消解紫外分光光度法测定总氮的过程中,过硫酸钾是至关重要的试剂。首先,试剂的纯度关系到空白值的高低、测定结果的准确度。一般普通分析纯过硫酸钾的总氮含量最高不超过0.005%,但由于试剂质量存在差异,有些厂家、批次的试剂含氮量常常达不到这个要求,致使空白值偏高。因此,有条件的话建议使用优级纯试剂,尽量降低试剂中的含氮量,从而降低实验空白值。5、实验用水及试剂的质量检验[b][/b]若实验的空白值不够理想,则需要对实验用水及试剂进行检验,以选择出含氮量最低的水和试剂,获得理想的空白值。[color=#ac1d10][b]粗蛋白测定空白偏高的影响因素?[/b][/color]可以从以下方面考虑:1.配溶液的水换没换;2.看看消化炉的回收率;3.看看蒸馏仪的回收率;4.盐酸在不在保质期内;5.配溶液的硫酸是不是有问题;6.检查催化剂,是不是含有硝酸钾。小结空白试验是实验室质量控制的重要环节,其准确性对提高检测结果的准确度至关重要。以分光光度法空白为例 ,要用相应的实验用纯水或有机溶济作参比,不能用空白实验溶液做参比,以免人为减小空白值,从而掩盖空白实验值的变异 。值得注意的是,通过计量认证的实验室,由于检测仪器和玻璃量器定期检定 ,以及采取了全面的质量管理措施,因此,若测定的空白实验值太大,一般是化学药品纯度不够,配制的试液变质或被污染等原因 。应重新配制试液 ,返工重测该批样品 ,直至空白实验值合格。
液相色谱的试验中,空白试样的色谱图怎样参与计算,是试样和空白试样的两个色谱图进行相减吗????????????
在蜂蜜糖分检测中,使用液相和示差,除了做平行试验还要做空白试验,如何做空白试验???????????[em26]
高效液相空白色谱柱出峰的原因? 高效液相色谱,色谱柱是Kromasil C18(15cm*5Um),流动相为10%乙腈和90%三氟乙酸水,走空白色谱柱时在整个保留时间的中间位置出现较大的色谱峰,走了好几针空白,每针的响应都不同,有的很高,有的又很低,但杂峰一直存在,不像是色谱柱里有杂质,用流动相冲洗后依然出现此问题,什么原因? 【分析原因】1.确定检测波长,如果是末端吸收,排除三氟乙酸的干扰。2.使用100%乙腈作为流动相,乙腈作为样品进样,如果为基线,则说明色谱柱及系统完好。3.然后在换流动相为10%乙腈和90%三氟乙酸水,样品为乙腈,如果有上述“中间位置出现较大的色谱峰”,那说明八成问题在三氟乙酸上,或者稀释三氟乙酸的水上。4.如果上述2、3都有“中间位置出现较大的色谱峰”,检查检测器及进样器。5.这些还不能解决,请联系工程师 空白乙腈出峰的原因? 高效液相色谱仪,进空白乙腈,在样品峰位子出现倒峰,进双蒸水在样品峰出现正峰,进样品时有杂峰干扰,我已经排除了水和乙腈的问题,不知道是不是进样池或者检测器污染了,已经整了10多天,还是没效果,请指教原因和解决办法。 【分析原因】液相中出现倒峰,主要原因是样品吸收比流动相吸收小,所以排除影响,不仅要考虑进样系统污染和检测池污染,还要考虑流动相和色谱柱的影响。换一根柱子试试,考察系统的情况,如果照旧,就排除色谱柱的影响,然后更换流动相试试,再以后清洗进样系统和检测池等等。 或将色谱柱从系统中断开,直接用乙腈或水做流动相清洗检测器就可以知道是否是检测器受污染,如果基线正常,再检查你的进样系统,样品,前处理是否有问题,如果都没问题,就去检查色谱柱吧。
工作初学液相色谱,领导要扣除空白样和积分,完全不懂,求大神指导
求实验室采购的用于微量、痕量物质色谱分析所用的有机试剂的空白试验操作作业指导书
新的C18液相色谱柱空白出峰是什么原因
一、自我介绍1、仪器:Agilent 12602、测试样品:纸张3、测试方法:YQ/T 35-2013《烟用纸张中甲醛和乙醛的测定 高效液相色谱法》:即以水萃取样品中的甲醛和乙醛,通过2,4-二硝基苯肼(即DNPH)衍生化生成甲醛苯腙,以高效液相色谱仪/紫外检测器测定,外标法定量。4、样品前处理:称取2g试样(纸),置于三角瓶,加入水,40°C浸泡24h,移取1mL上清液转置10mL容量瓶,加入DNPH衍生化试剂,30min,过0.45微米有机膜后,上机分析。5、标准溶液:称取甲醛-苯腙标样,溶于乙腈,按比例稀释至系列标准工作溶液,对应甲醛浓度依次为0.014mg/L, 0.028mg/L, 0.071mg/L, 0.142mg/L, 0.569mg/L。6、仪器条件:(1)C18反相色谱柱,(2)流动相A,水(3)流动相B,乙腈(4)柱温30°C(5)柱流量,1.5mL/min(6)进样体积,10微升(7)梯度洗脱20min。0-10min: 50%水+50%乙腈。13-18min: 25%水+75%乙腈。18-20min: 50%水+50%乙腈。(8)紫外检测器=======================================绝望地分割===========================================二、病状描述 由于是以水萃取样品中的甲醛,所以每次都带空白样(即水)。 计算公式:甲醛含量=样品测定值-空白测定值。 关键是空白值老是很高,例1:空白值=0.0953mg/L,测定值=0.1179mg/L。T.T结果误差可想而知了。 想来第一反应应该是水有问题! 水为纯水机制备的一级水,这个一级水在纯化过程中对甲醛的去除率究竟如何? 最近试了3个水,a纯水机超纯水,b娃哈哈纯净水,c工厂冷凝水。结果发现甲醛浓度一如既往地高,而且三者浓度如此接近! 想来第二反应是不是那儿有污染源?污染浓度远高于3个水的浓度,以至于测定时三者如此接近? 唠唠叨叨的,敢问大神怎么解决这个空白值高的问题?????????????
[url=https://insevent.instrument.com.cn/t/5p][color=#3333ff]液相色谱[/color][/url]进一针空白出来这个谱图,是什么原因?[img]https://ng1.17img.cn/bbsfiles/images/2022/12/202212031458422286_9904_5889169_3.png[/img]
液相色谱经常在空白和低浓度对照溶液中出现异常峰,不同仪器不同色谱柱,不同产品,都是经常出现不规则鼓包峰,求助各位大神
[url=https://insevent.instrument.com.cn/t/5p][color=#3333ff]液相色谱[/color][/url]空白溶液总是出正负峰是什么原因导致的
液相色谱,走空白走不好,太多杂峰,但是同样的柱子和条件在别的液相走的很好,是什么原因呢?
[color=#444444]使用液相色谱,流动相是20mmol磷酸二氢钠,流速0.5,进样流动相,或者样品,标准品,在5.1min左右都有一个杂峰,大小不定,没有规律,并且 只切换阀,不进样峰就会变小,这个是什么原因呀,是仪器问题,还是样品问题?[/color]
各位老师:我的液相色谱,流动相识甲醇:乙腈=7:3,基线很好,进溶剂及空白针都很正常,但是只要一进样品,就会在同样的位置出鬼峰,而且每针样品都出同样的大小不停变化的鬼峰,请教各位老师,这究竟是什么情况?
有哪位大神可以帮我解答一下,日立液相色谱(荧光检测器和紫外检测器)测定多环芳烃时空白基线是平的,跑标样时基线梯度往上升,而且多环芳烃最后两种物质同一种方法以前可以分开,现在分不开了,是什么原因?
求实验室采购的用于微量、痕量物质色谱分析所用的有机试剂的空白试验操作作业指导书
液相色谱总是有杂峰,走空白针也是有杂峰,之前没有这个杂峰,最近才有的。流动相是乙腈和水,用的多环芳烃专用柱子。流动相换过了,装流动相的瓶子也洗过了,用异丙醇洗柱子洗了一宿,但是进空白针还是有杂峰且杂峰没变化,求大神指导!
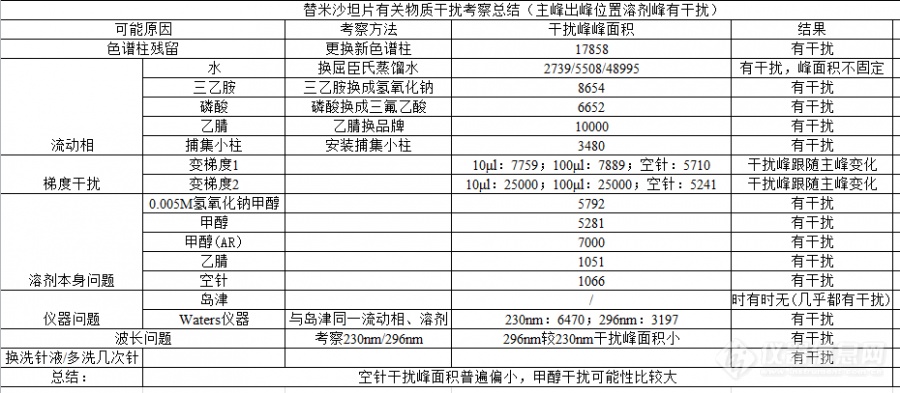
[color=#444444]高效液相色谱法空白溶剂总是干扰主峰检测,排除方法见下[/color][color=#444444][img=,690,301]https://ng1.17img.cn/bbsfiles/images/2019/06/201906210958176242_7049_1827556_3.png!w690x301.jpg[/img][/color]
请问各位大神,做高效液相色谱实验中需要配制溶液,溶液的配制过程中有哪些操作的关键步骤?
[color=#444444]最近做除虫脲和灭幼脲液相色谱标样分离,色谱条件甲醇-水=75+25,流速1mL/min,检测波长260nm,单标样的峰分裂了,之前做过,峰形很好,怀疑柱子污染了,换了新的柱子还是一样,预柱去了也还是分不好,除虫脲和灭幼脲性质稳定用的乙腈溶解的,不知道什么原因造成的,进过空白溶剂的峰,不会产生杂峰。[/color]
[color=#444444]液相色谱做迁移试验模拟物加标回收率,不懂模拟物加标,是空白加标回收吗,那添加0.1mg/l这是个浓度,具体加多少体积呢,是自己定吗[/color]
请教一下,使用高效液相色谱对于试验环境有什么要求?是否有什么法规或者标准、通则中有提到。
各位高手:在液相色谱中,如果在空白峰保留时间前出现有特征紫外吸收的峰,怎么办?是直接算杂质?还是调整色谱条件,让此峰在此条件下有保留,怎么调整色谱条件?
我对液相色谱实验有这样一个体会:入门容易,成为高手难。做过实验的都知道,对于简单样品、成熟方法,HPLC很方便,尤其是有了自动进样器以后,做实验倒是还方便些,大量的时间都在样品前处理和数据处理计算上。但是偶尔,会碰到让人心烦的事情。比如,做间苯三酚的有关物质实验。用的柱子是Zorbax C18,250*4.6mm,5μ.Agilent 1100,UV检测器 流速:1.0ml/min流动相乙腈-水(用磷酸调pH到3.0)(10:90)配制样品的溶剂就是流动相。结果空白进样后,7.6min出来一个小峰,峰面积在30左右,再进一针,只有25了,再进一针,没有了!样品溶液两个浓度进样,其他位置的峰重现性极佳,就是7.6min出的峰忽大忽小,后来干脆全部重新进样,终于稳定了。碰到这样的问题,我的解决办法是这样,从理论上去判断,首先空白溶液在7.6min不应该出峰。那种异常现象没有时间去多想,否则会发疯的!一个培训的老师说过,不是每个人都能成为科学家的,更多的人只要学会应用就可以了。样品溶液的异常情况也没有更好的解决办法,只能多进几针,取重现性好的。写在这里,欢迎大家交流实验中出现的异常个例。
我单位要建立一个高效液相色谱实验室,由于没有招标的资金,所以自己设计。请问,建立高效液相色谱实验室应考虑哪些方面的问题?如何解决?[em07]
[color=#444444]我做液相色谱的分离度实验。药典要求将样品酸化2小时后,加碱回调ph,这样能得到主峰的水解产物并且RRT=0.9。但是最后得出的结果并没有这个峰。并且,其他杂质反而变大了,主峰也没有降解的迹象。希望老师能帮忙解答我的难题。[/color]
问题: 请教:液相色谱实验中,流动相对柱子压力有什么影响呢回复: 粘度影响压力,流速相同的情况下
[color=#444444]我要做高效液相色谱的实验,涉及到买标准品,国外很多文章不承认国内的标准品,打算买国外的,请问怎么才能查到我所买公司的标准品被国外文章承认与否?[/color]
[color=#444444]现在用液相色谱测定甲酸,但是,甲酸在C18柱子里基本没有保留,总是与空白的峰无法分开。求帮助[/color]
[url=https://insevent.instrument.com.cn/t/5p][color=#3333ff]液相色谱[/color][/url]出峰问题:流动相为乙腈:甲醇:纯水我们在测样品时发现保留时间在七分钟一直有一个峰,走空白也有这个峰,同样的色谱条件,我们去QC拿了一个样品回来检测,发现七分钟这个峰没有了,但是我们其他样品检测时都有这个峰,怎么判断这个峰是不是属于空白峰呢?如果是空白峰为什么检测别人配制的样品就没有呢?







 我要推广仪器
我要推广仪器
 下载APP
下载APP