用液相色谱测定化合物的含量时,没有标准品,目前只能用面积归一化来定量,我感觉这样很不准确,大家有什么好的办法没有?
[color=#444444]液相色谱做定量时,如果标准品和样品称量一样,进样量也一样,是不是说,物质的量之比就等于峰面积之比呢?[/color]
关于液相色谱的定量分析定量分析是在定性分析的基础上,需要纯物质作为标准样品。液相色谱的定量是相对的定量方法,即:由已知的标准样品推算出被测样品的量。
液相色谱仪进行空白溶液注射后悔产生与前一样品或标准溶液无关的峰。冲洗针管并采用相同方式注射空白溶液。1 如果峰消失,说明针管被污染,冲洗已将其清洁,今后需要注意对针管进行定期例行冲洗。2 如果峰仍然存在,冲洗,切换至取样位置,立即进行并注射相当于定量环体积1/4的空白溶液,观察峰。再冲洗,切换至取样位置,立即进样并注射以定量环体积5倍的空白溶液。观察以下峰的变化。①如果第二次注射峰比第一次的大3~4倍,判断是来自注射器和玻璃器皿的外部污染。解决办法是使用干净的器具。不要使用会产生污染物的塑料,进一步确认鬼峰是否为氧气峰。②如果第二次注射峰消失,或者比第一次小了很多,证明是液相色谱仪进样阀内的污染物。解决办法是清洗进样阀。但有时采用其他解决方法更为方便。如果使用的是不分充填的方法,可以改用完全充填,并在必要时使用体积较小的定量环。③第二次注射的峰与第一次相同,仅仅在进行取样与进样之间而得相互切换就可使注射器内的微量污染物被清洗。确认方法:在取样位置进行冲洗,清洁放空管。切换至进样位置等待鬼峰出现之后,进行正常冲洗,在任然处于进样位置的状态下,插入一根非常干净的空注射器并保持其位置不变,以防止放空管内残留污染物或针密封上的污染物因虹吸或扩散进入定量环。将手柄切换至取样,无须再切换至进样,气动记录器。等待洗脱出的波峰。对峰进行观察。然后,注射器不动,将手柄切换至进样,再次气动记录器,观察峰。这两次操作中,只要有峰产生,就说明有内部污染物,且多数发生在转子密封圈上。这样的情况需要清洁进样阀。
岛津的液相色谱,配有自动进样器。用外标定量时,只配一个标样,选择用改变的进样量(即1微升,2微升。。。。)这种方式来做标准曲线,是不是线性更好一些。
各位前辈,请教一个问题,关于液相色谱定量时对于有检出的阳性样品复测是否一定要走标液曲线,如果用单点定量的检测值的准确度是否会与标液曲线定量的值相差较大;对于阳性样检出值接近检测限的情况两者哪个更有优势,毕竟走单点可以节省很多时间。。。。

在液相色谱分析过程中,有时候客户经常会遇到色谱峰展宽的情况,有时候是系统方面的原因,一般可以解决;有时候是色谱柱受到了污染或者损坏,小的问题,通过修补可以缓解,如果柱子损坏比较严重,可能就无力回天了~~以下是柱性能下降的各种可能情况,及解决办法:柱性能下降的一个重要现象就是在保留时间基本不发生变化的情况下,色谱峰的区域宽度明显增大(如下图)。http://ng1.17img.cn/bbsfiles/images/2017/10/2015041009461674_01_2452211_3.jpghttp://ng1.17img.cn/bbsfiles/images/2017/10/2015041009462350_01_2452211_3.jpg死体积:从进样器到检测器间流动相充满的体积,它包括进样器定量环体积、管路体积、色谱柱填料间隙体积、流通池体积。为了抑制色谱峰区域展宽,死体积应尽可能的小具体做法如下:(1)进样器:在分析型液相色谱中,定量环体积从5 μL~100 μL不等;日常分析应配备多款体积不等的定量环,分析时选择比进样体积大但与进样体积最接近的那一种。(2)管路:从进样器到流通池间的管路,内径应小于0.01英寸,长度则应尽量短;其他部位的管路的内径可以大一些。(3)流通池:对于长度超过150mm,内径超过4mm的常规柱,可以使用常规流通池(8~10μL);规格较小的短柱和细径柱,则应使用半微量(~5μL)和微量流通池(~2μL)。http://ng1.17img.cn/bbsfiles/images/2017/10/2015041009462389_01_2452211_3.jpg
[color=#444444]最近在用液相色谱仪做水杨酸的定量,水杨酸用甲醇:水=60:40配制,流动相为甲醇:水(0.01M甲酸铵)=60:40,但是不知道什么原因,水杨酸的面积随着时间的推移一直在变小?另外,问下大家是用什么方法做的[/color]
液相色谱工作中和色谱柱有关的故障及解决办法1.保留时间漂移A 温度变化 设定一个恒定的温度,而且当室温较所需温度低时要注意提高柱温的设定值。B 流动相的问题使用预混合则要注意每次配液的准确性、精密度与pH值的变化;流动相被污染或加入添加剂后可能对待测组分存在干扰。流动相的pH值和样品的pK值太接近,即便是使用有缓冲盐的流动相也会引起保留时间的波动,流动相的pH值比待分离组分的pK值至少相差2个pH单位2.异常的色谱峰A 出现一个或几个负峰 流动相吸收本底高;进样过程中进入空气;由于样品中的某个组分的紫外吸收低于流动相的紫外吸收。B 均为负峰信号电缆接反或检测器输出极性设置颠倒;光学装置尚未达到平衡。C 均为宽峰系统未达到平衡;溶解样品的溶剂极性在于流动相极性;色谱柱尺寸及类型选择不正确;色谱柱或保护柱被污染或柱效降低;由于温度变化所产生样品粘度过大;进样器故障或进样体积误差;检测器设置不正确,定量环体积不正确;检测池污染;检测器灯出现故障。D 出现双峰或肩峰进样量或样品浓度过高;溶解样品的溶剂极性较流动相极性强;保护柱或色谱柱污染或失效或堵塞;过滤器堵塞;分析柱“柱头塌陷”。E 拖尾峰检测器设置不正确;进样体积太大或样品浓度太高。较早流出的峰拖尾,柱外效应引起的(检查液相色谱系统的毛细连接、毛细管及检测池);较晚流出的峰拖尾,待分离化合物和固定相表面非特异性相互作用(在流动相中加入三乙胺或醋酸盐或者选择合适的固定相可以改善)F 出现平头峰出现鬼峰可能为上次样品的残余。在每次进完样后需要用充足的时间来平衡和清洗整个系统;样品中存在未知物,改进样品的预处理;流动相污染,更换新流动相,尽可能做到流动相现配现,隔夜的流动相应放入冰箱储存再次使用时要进行过滤,尽可能使用HPLC级试剂。贴心提示: ①保留时间没有规律的变化说明色谱柱没有充分平衡。随着色谱柱使用时间的增加,保留时间会前移,特别是当流动相的pH值为酸性时(pH≤2)。如果突然发生明显变化,一般由于系统的问题。②以上的问题只是工作中常见的部分问题,如有未提及的问题出现,请咨询您的供应商或厂家技术服务人员。③液相色谱故障排除时不应以问题的表征消失而告终,应该着重于在众多可能的因素中找出问题的根源、每次仅改变一个因素,系统地将问题定位,直至最终筛选出真正产生故障的因素④将换下来的有问题的部件扔掉,以免和其他好的混淆。保留的旧部件请用标签做好标记。⑤实验室应该具有优良的预防性的维护措施合严格周密的记录。对于常规分析,我们应该大致了解一根柱子/预柱可以进样多少针,这样可以将问题发生的频率最小化。
液相色谱如何进行定性和定量分析?
请问许多文献上提出核磁内标法(绝对测量法)定量分析时不需引进任何校正因子,不需制作标准曲线,但高效液相色谱为何需制作标准曲线?高效液相色谱和核磁内标法定量的原理差不多呀?[em09]
[color=#00008B][B]该帖为楼主自己整理,帖中所有楼主撰写的内容未经授权不得转载,否则属侵权违法行为![/B][/color]看到很多版友发帖求助讨论准确性的问题,我特意整理了一个关于定量分析误差问题的帖子,希望对大家有所帮助。高效液相色谱定量分析过程可分为样品的前处理、标准品的配制、进样、色谱分离、检测及数据处理等七个步骤。[B]一 误差的主要来源[/B]随着现在市场销售仪器自动化程度的提高,进样、色谱分离、检测及数据处理等实验环节对实验结果产生的误差越来越小,尤其在高效液相色谱定量分析中,实验结果的误差可能主要来源于样品的前处理及标准品的配制。[B]1、样品的前处理 [/B]样品的萃取率是样品前处理时存在的主要问题。当固-液萃取(含柱分离的前处理方法),液-液萃取时,存在萃取率不高且不稳定的问题。尤其在除去蛋白质时,存在变性蛋白质会吸附一些被测组分,导致萃取率降低的情况。通常,萃取率是通过在式样中添加被测成分在萃取的方法评价的。也就是说,被测成分的增加量和液相色谱中的峰面增大成比例关系,通过这种方法可以确定溶液中被测成分的变化趋势。 如果存在萃取率不稳定的问题,就有必要改变萃取的方法,要预先添加内标,然后萃取。这种情况采用的内标,必须与被测物质的化学结构类似,萃取的萃取率才可能相近。如果回收率不但接近100%而且较为稳定,可以证明这种前处理方法较为可靠。在分析中如果能充分考虑以上误差产生的各种原因,才有可能得到精确的分析结果。 [B]2、标准品的配制[/B]本帖中讨论的问题带有普遍性,影响高效液相色谱分析结果准确性的因素较多,仅在标准溶液的配置过程中,可以分为标准物质的称量,溶液的配制和溶液的储存三个环节。 作为标准溶液使用的标准物质其纯度要求很高,应避免使用纯度不符合要求的试剂,作为标准溶液使用的标准物质具有不可替代性,现在市场有销售的HPLC专用的溶剂及各种标准试剂可供实验选择;其次选择与样品浓度要求相适应的天平,应该尽可能使用高精度的天平,这样才能把由于天平使用带来的称量操作误差降至最低。[B]二 消除误差的方法[/B] 要提高分析结果的准确度,必须考虑在分析过程中可能产生的各种误差,采取有效措施,将这些误差减到最小。[B]1、选择合适的分析方法[/B] 各种分析方法的准确度是不同的。化学分析法对高含量组分的测定能获得准确和较满意的结果,相对误差一般在千分之几。而对低含量组分的测定,化学分析法就达不到这个要求。仪器分析法虽然误差较大,但是由于灵敏度高,可以测出低含量组分。在选择分析方法时,一定要根据组分含量及对准确度的要求,在可能条件下选最佳分析方法。[B]2、增加平行测定的次数[/B] 如前所述增加测定次数可以减少随机误差。在一般分析工作中,测定次数为2—4次。如果没有意外误差发生,基本上可以得到比较准确的分析结果。[B]3、消除测定中的系统误差[/B] 消除测定中系统误差可采取以下措施:其一是做空白实验,即在不加试样的情况下,按试样分析规程在同样操作条件下进行的分析。所得结果的数值称为空白值。然后从试样结果中扣除空白值就得到比较可靠的分析结果。其二是注意仪器校正,具有准确体积的和质量的仪器,如滴定管、移液管、容量瓶和分析天平,都应进行校正,以消除仪器不准所引起的系统误差。因为这些测量数据都是参加分析结果计算的。其三是作对照试验,对照试验就是用同样的分析方法在同样的条件下,用标样代替试样进行的平行测定。将对照试验的测定结果与标样的已知含量相比,其比值称为校正系数。 校正系数=标准试样组分的标准含量/标准试样测定的含量 被测试样的组分含量=测得含量×校正系数 综上所述,在分析过程中检查有无系统误差存在,作对照试验是最有效的办法。通过对照试验可以校正测试结果,消除系统误差。[B]4、样品定量分析过程中的误差[/B]样品处理要尽量减少操作者的技术问题带来的误差,样品的稀释次数、稀释工具都是误差的祸根,应尽量减少稀释次数,稀释工具用高准确度的。样品中的干扰组分会直接影响分析的准确度,而且有些组分会损坏柱子。纯化样品的过程尽量少用蒸发至干的步骤(在色谱分析中这一步又是不可少的),正确操作固相柱萃取、纯化小柱使用的步骤,注意提高每一步的回收率,使用内标法也是一个能准确定量的方法。 在手动进样中进样体积至少是样品定量环管体积的3倍,色谱分离程序要使色谱峰的分离度大于1.5,控制流动相、流量、温度等的平稳。流动相的污染都会抬高基线或减少信噪比,分辨率下降,试验条件的变化,如柱退化、不好的流动相等都能引起保留时间变化,引起一个峰或更多的峰不能被鉴别。正确设定仪器参数,选用合理的数据处理参数,用峰面积计算结果比峰高更精确。[B]三 结论 [/B]以上的分析的是我们能尽量控制的误差,还有一些不是操作者所能控制的误差,如被测定组分易分解、组分的含量高低、介质效应等。我们把能控制的误差减小到最低,那你的结果准确度将更高。
高效液相色谱串联质谱可以定量吗?
液相色谱法定量用外标法定量时,含量超过100%的可能性有多少,可信吗?有多少改善的余地?
有什么办法能让单泵液相色谱进行梯度洗脱?大家讨论下[em52] 我想到一个办法,不知道行不行但是现在还没想好[em53]
各位,液相色谱定量优于气质,具体原因要如何解释?给人培训,要比较专业的术语来解释,一下把我难到了
液相色谱分析间苯二磺酸和苯磺酸的条件及定量方法?
前几天做的实验能用液相色谱能把饱和分、芳香分和胶质分开,但只是知道各部分出的峰的面积,因为饱和分、芳香分和胶质都不是纯物质,而且不同物质的同一组分性质又不一定相同,所以无法知道每一组分的校正因子,不知道如何定量,请高手帮忙!
各位大神,实在木有办法,对这一片太不熟悉了,能帮我看看这道题吗?一台高效液相色谱进样环为20ul,做定量分析,一次进样为26ul,另一次进样为18ul,这两次进样对分析结果分别有什么影响
硼砂做液相色谱,外标定量,峰面积相差不大,
[color=#444444]液相色谱进行简单的峰面积比定量分析时,如果合成的样品的溶剂是四氢呋喃,而标准品的溶剂是甲醇,而且合成的样品产量很少,请问如何将四氢呋喃转换成甲醇?是否可以通过氮气吹干四氢呋喃溶剂;是否还有别的可行的方法?[/color]
各位大侠; 我现在使用的液相色谱定量方法是在建立方法时坐标准曲线,在以后测定样品含量时就一直套用以前的标准曲线求值,现在问题是怎么确保曲线的可用性,系统的稳定性一直好可以一直用以前的曲线求值吗?
液相色谱出峰拖尾比较严重,想知道造成这种现象的原因及相对应的处理办法,多谢
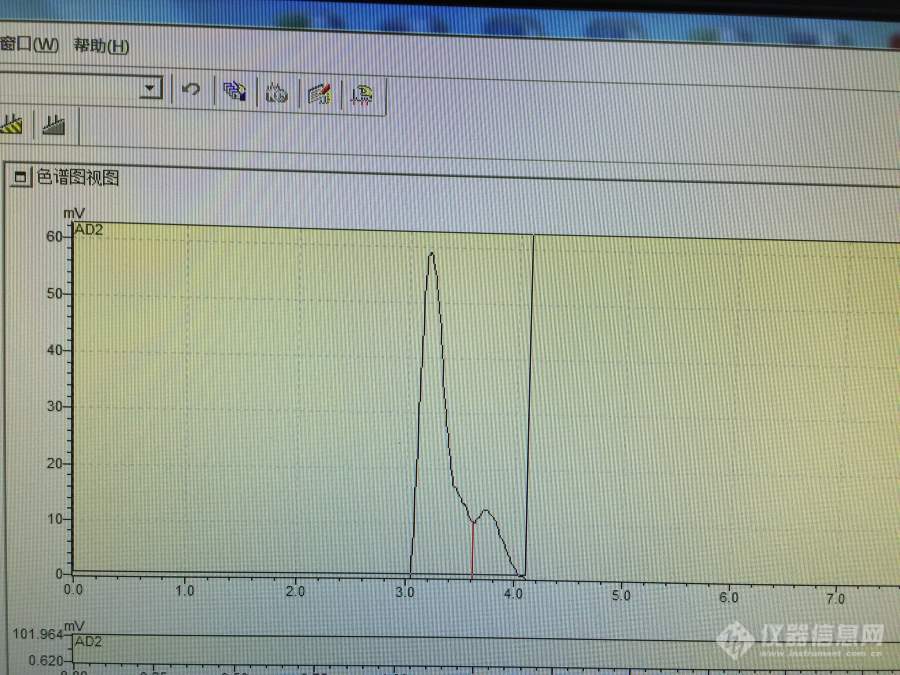
[color=#444444]使用蒸发光检测器的液相色谱测定丁二酸和戊二酸混合物中两组分含量,流动相是甲醇-水,改变过流动相配比,但是仍然分不开。看过文献,有用示差折光率检测器分开这两种物质的,在流动相中加了乙酸乙酯。请问各位牛人,如果用蒸发光检测器,有什么解决办法能分开这两种物质吗?先谢过各位了。[/color][color=#444444][img=,690,517]https://ng1.17img.cn/bbsfiles/images/2019/06/201906131108182091_1175_1827556_3.jpg!w690x517.jpg[/img][/color]
液相色谱和[url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]的外标法和内标定量有何区别?是如何处理的?
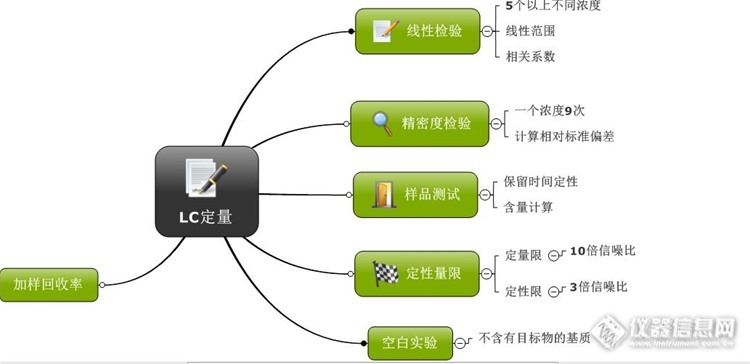
今天读到傅若农教授的文字,感到很温暖。想想我这个分析化学战线的逃兵,将彻底地和分析化学说再见了。但是,我还是讲我的一些思路做一个总结,希望能给我们90后的分析工作者有点启发。接着去年我那个帖子。《谈谈液相色谱方法开发》,有网友希望我写点定量方面的内容。那么,今天,我权当抛砖引玉。当然,我写的很不成熟,希望大家批评指正。http://bbs.instrument.com.cn/topic/5957822在做好了分离方法之后,我们需要对我们的目标物进行定量分析。定量分析的思路一般是http://ng1.17img.cn/bbsfiles/images/2016/06/201606241414_597987_1626663_3.png一般来说,通过这套思路来进行定量方法学的验证。这个是一个通用的方法。当然,这也是十五多年前我做课题时候。 我导师教我的,现在也许也有发展了吧。
请教各位前辈关于用液相色谱检测甲醛的方法,目前我是用2,4-二硝基苯肼与甲醛反应后用二氯甲烷萃取,但发现结果试剂空白(纯水)中甲醛的含量也不低,导致标液定量时比较麻烦,不知道有做过的前辈是否有类似的情况,有什么好的解决办法吗,麻烦知道的前辈多多指点,本人不胜感激!!!
跟大家分享一下液相色谱讲义,里面包括(1)液相色谱基础知识(2)液相色谱的方法开发-分离机理及色谱柱(3)分离方法,离子抑制及离子对方法介绍(4)梯度方法的开发及梯度分离时的注意事项(5)液相色谱的实用技术,色谱柱的保养、样品前处理(6)液相色谱的定性、定量分析(7)色谱泵和进样器的原理及操作(8)检测器的原理及操作
[url=https://insevent.instrument.com.cn/t/5p][color=#3333ff]液相色谱[/color][/url]主要用于对吸收紫外线的液态有机物(不饱和有机物)进行定性、定量分析和纯度分析,定性,是保留时间的特征峰值。没有办法分析物质结构,但如果已知的物质是x,则要测量的物质可以与x标准物质比较。[url=https://insevent.instrument.com.cn/t/5p][color=#3333ff]液相色谱[/color][/url]测定表明,该物质的停留时间相同,测定结果表明,该物质主要用于测试中的色谱鉴定
液相色谱流动相中有缓冲盐,系统总爱进气泡,大家有没有好的解决办法?附:缓冲盐是磷酸氢二钾和磷酸二氢钾的水溶液,pH=6.8,流动相是60%的乙腈+40%的盐。谢谢大家了~







 我要推广仪器
我要推广仪器
 下载APP
下载APP