新型蛋白质结构分析手段-氢氘交换质谱技术进展
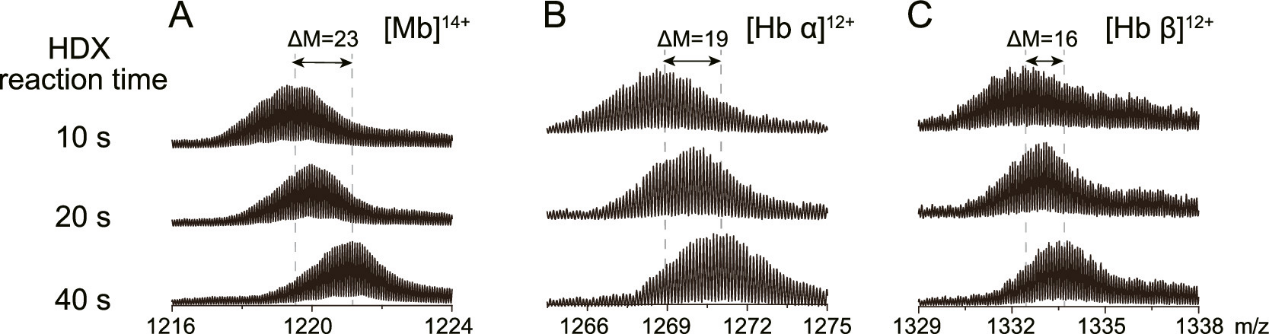
贾伟、陈熙沃特世科技(上海)有限公司实验中心氢氘交换质谱法是一种研究蛋白质空间构象的质谱技术。它在蛋白质结构及动态变化研究、蛋白质相互作用位点发现、蛋白表位及活性位点鉴定方面有着广泛的应用。随着氢氘交换质谱技术的不断发展,它正在成为结构生物学家及生物药物研发的重要手段。 氢氘交换质谱(HDX MS,hydrogen deuterium exchange mass spectrometry)是一种研究蛋白质空间构象的质谱技术。其原理是将蛋白浸入重水溶液中,蛋白的氢原子将于重水的氘原子发生交换,而且蛋白质表面与重水密切接触的氢比位于蛋白质内部的或参与氢键形成的氢的交换速率快,进而通过质谱检测确定蛋白质不同序列片段的氢氘交换速率,从而得出蛋白质空间结构信息[1]。这个过程就像将握着的拳头浸入水中,然后提出水面并张开手掌。这时,湿润的手背表明它在&ldquo 拳头&rdquo 的结构中处于外表面,而较为干燥的手心表明它是&ldquo 拳头&rdquo 的内部。除样品制备外,氢氘交换质谱法的主要过程包括:交换反应、终止反应、将蛋白快速酶切为多肽、液相分离、质谱检测、数据解析。其中交换步骤需要在多个反应时长下进行,如0s、10s、1min、10min、60min等,以绘制交换率曲线,得到准确全面的信息。氢氘交换质谱技术在蛋白质结构及其动态变化研究[1]、蛋白质相互作用位点发现[2]、蛋白表位及活性位点鉴定方面有着广泛的应用[3]。 与经典的蛋白质结构研究方法相比,如X射线晶体衍射(X-Ray Crystallography)和核磁共振(NMR. Nuclear Magnetic Resonance)等方法,氢氘交换质谱不能够提供精确的蛋白空间结构,它直接提供的主要信息包括哪些氨基酸序列位于蛋白质空间结构的表面位置(包括动态变化中的)、可能的活性位点和蛋白-蛋白相互作用位点等。但是氢氘交换质谱技术有着其他经典方法不具备的优点:首先,可以进行蛋白质结构动态变化的研究是氢氘交换质谱的一个突出优点,包括变化中的活性位点及表位;其次,氢氘交换质谱在蛋白复合体构象的研究中也具有独到的优势;此外,氢氘交换质谱还具有对样品需求量小、纯度要求相对较低、研究对象为溶液环境下的蛋白质的天然构象而非晶体中构象等优势[1,4,5]。自1991年第一篇研究论文发表起,氢氘交换质谱技术不断发展,已经成为结构生物学及质谱技术中一个非常重要的应用领域[6]。但是氢氘交换质谱实验的复杂的实现过程在一定程度上影响了其应用的广泛度。主要的难点有:1、如何避免交换后氘代肽段的回交现象;2、实验控制的高精确性和重现性要求;3、交换后造成的叠加的质谱峰如何准确分辨;4、简易高效的分析软件需求;5、以氨基酸为单位的交换位点辨析。沃特世公司自2005年起,针对以上难点不断进行攻关,推出了目前唯一商业化的全自动氢氘交换质谱系统解决方案&mdash &mdash nanoACQUITY UPLC HD-Exchange System(图1)。在全世界范围内,这套系统已经帮助科学家在包括Cell、Nature等顶级研究期刊中发表研究论文[7,8]。除科研需求外,沃特世氢氘交换质谱系统也受到众多国际领先制药公司的认可,并用于新药开发中蛋白药物活性位点及表位的研究工作中。氢氘交换实验中的回交现象将严重影响实验数据的可信度,甚至导致错误结果的产生。要避免回交需要做到两点:尽量缩短液质分析时间和保证液质分析中的温度和pH为最低回交反应系数所要求的环境。沃特世UPLC系统采用亚二纳米色谱颗粒填料,较HPLC使用的大颗粒填料,UPLC具有无与伦比的分离度。因此UPLC可以做到在不损失色谱分离效果的要求下,极大缩短液相分析时间的要求[9]。对于对温度和pH控制问题,在多年的工程学改进中,nanoACQUITY UPLC HD-Exchange System已经实现了对酶切、液相分离等步骤的全程控制[10]。 对氢氘交换质谱实验精确性和重现性的要求是其应用的第二个主要难点。在实验中一般需要采集0s、10s、1min、10min、60min、240min等多个时间点的数据。如果进行人工手动实验,很难做到对10S-10min等几个时间点的精确操作。再考虑到重复实验的需求,人工手动操作会对最终数据可信度产生影响。而且实验过程重复繁琐,将给实验人员带来非常大的工作压力。nanoACQUITY UPLC HD-Exchange System完全通过智能机械臂,精确完成交换、终止交换、进样、酶切等一系列实验过程,而且始终保证各个步骤所需不同的温度环境。这些自动化过程不但保证了实验数据的可靠性,提高了实验效率,也将科学家从繁琐的重复实验中解放出来。 氢氘交换实验的质谱数据中,随着交换时间的延长,发生了交换反应的多肽,由于质量变大,其质谱信号将逐渐向高质荷比方向移动。因此,这些质谱峰可能与哪些未发生交换反应的多肽质谱峰逐渐叠加、相互覆盖。相互叠加的质谱信号,不但影响对峰归属的判断,更会增加交换率数据的误差。因为交换率判断需要通过对发生交换的多肽进行定量,毫无疑问因叠加的而混乱的质谱数据将极大的影响对质谱峰的准确定量。这点对于单纯通过质荷比进行分析的质谱仪来说完全无能为力。但是,这个看似不可能完成的任务却被沃特世 nanoACQUITY UPLC HD-Exchange System攻克了。这是因为,不同于其它常见质谱,沃特世的SYNAPT质谱平台还具备根据离子大小及形态进行分离的功能(行波离子淌度分离)。在数据处理时,除多肽离子的质荷比信息外,还可以通过离子迁移时间(离子淌度维度参数)将不同离子区分。因此这种SYNPAT独有的被命名为HDMSE的质谱分析技术可以将因质荷比相同而重叠的多肽分离开,轻而易举地解决了质谱信号叠加的问题,得到准确的交换率数据[11,12](图2)。SYNPAT质谱平台一经推出就夺得了2007年PITTCON金奖,目前已经推出了新一代的SYNAPT G2HDMS、SYNAPT G2-S HDMS等型号,并具备ESI、MALDI等多种离子源。除氢氘交换技术外,SYNAPT质谱系统在蛋白质复合体结构研究中也是独具特色,已有多篇高质量应用文献发表[13,14,15]。 实现氢氘交换质谱技术的第四个关键点,是如何高效分析实验产生的多时间点及多次重复带来的大量数据。人工完成如此巨大的信息处理工作,将消耗科学家大量的时间。沃特世氢氘交换质谱解决方案所提供的DynamX软件可以为科学家提供简便直观的分析结果,并包含多种呈现方式。 在某些特殊研究中,要求对蛋白氢氘交换位点做到精确到氨基酸的测量,这是氢氘交换质谱研究的又一个难点。在常规的研究中采用CID(碰撞诱导解离)碎裂模式,可能导致氘原子在多肽内重排,而致使不能对发生交换的具体氨基酸进行精确定位。SYNPAT质谱提供的ETD(电子转移解离)碎裂模式可以避免氘原子重排造成的信息混乱,并具有良好的碎裂信号[16]。沃特世的nanoACQUITY UPLC HD-Exchange System为氢氘交换质谱实验提供了前所未有的简易的解决方案,强有力地推动了氢氘交换技术在蛋白质结构及动态变化研究、蛋白质相互作用位点发现、蛋白表位以及活性位点鉴定方面的应用,正在成为众多结构生物学科学家和生物制药企业必不可少的工作平台。参考文献(1) John R. Engen, Analysis of Protein Conformation and Dynamics by Hydrogen/Deuterium Exchange MS. Anal. Chem. 2009,81, 7870&ndash 7875(2) Engen et al. probing protein interactions using HD exchange ms in ms of protein interactions. Edited by Downard, John Wiley & Sons, Inc. 2007, 45-61(3) Tiyanont K, Wales TE, Aste-Amezaga M, et al. Evidence for increased exposure of the Notch1 metalloprotease cleavage site upon conversion to an activated conformation. Structure. 2011, 19, 546-554(4) Heck AJ. Native mass spectrometry: a bridge between interactomics and structural biology. Nat Methods. 2008, 5, 927-933.(5) Esther van Duijn, Albert J.R. Heck. Mass spectrometric analysis of intact macromolecular chaperone complexes. Drug Discovery Today. Drug Discovery Today: Technologies Volume 3, 2006, 21-27(6) Viswanat ham Katta, Brian T. C hait, Steven Ca r r. Conformational changes in proteins probed by hydrogen-exchange electrospray-ionization mass spectrometry. Rapid Commun. Mass Spectrom. 1991, 5, 214&ndash 217(7) Chakraborty K, Chatila M, Sinha J, et al. Chaperonin-catalyzed rescue of kinetically trapped states in protein folding. Cell. 2010 Jul 9 142(1):112-22.(8) Zhang J, Adriá n FJ, Jahnke W, et al. Targeting Bcr-Abl by combining allosteric with AT P-binding-site inhibitors. Nature. 2010,463, 501-506(9) Wu Y, Engen JR, Hobbins WB. Ultra performance liquid chromatography (UPLC) further improves hydrogen/deuterium exchange mass spectrometry. J Am Soc Mass Spectrom. 2006 , 17, 163-167(10) Wales T E, Fadgen KE, Gerhardt GC, Engen JR. High-speedand high-resolution UPLC separation at zero degrees Celsius. Anal Chem. 2008, 80, 6815-6820(11) Giles K, Pringle SD, Worthington KR, et al. Applications of a travelling wave-based radio-frequency-only stacked ring ion guide. Rapid Commun Mass Spectrom. 2004, 18, 2401-2414(12) Olivova P, C hen W, C ha kra borty AB, Gebler JC. Determination of N-glycosylation sites and site heterogeneity in a monoclonal antibody by electrospray quadrupole ion-mobility time-offlight mass spectrometry. Rapid Commun Mass Spectrom. 2008, 22,29-40(13) Ruotolo BT, Benesch JL, Sandercock AM, et al. Ion mobilitymass spectrometry analysis of large protein complexes. Nat Protoc.2008, 3, 1139-52.(14) Uetrecht C, Barbu IM, Shoemaker GK, et al. Interrogating viral capsid assembly with ion mobility-mass spectrometry. Nat Chem.2011, 3,126-132(15) Bleiholder C, Dupuis NF, Wyttenbac h T, Bowers MT. Ion mobility-mass spectrometry reveals a conformational conversion from random assembly to &beta -sheet in amyloid fibril formation. Nat Chem.2011, 3, 172-177(16) Kasper D. Rand, Steven D. Pringle, Michael Morris, John R., et al. ETD in a Traveling Wave Ion Guide at Tuned Z-Spray Ion Source Conditions Allows for Site-Specific Hydrogen/Deuterium Exchange Measurements. J Am Soc Mass Spectrom. 2011, in press





















 我要推广仪器
我要推广仪器
 下载APP
下载APP