[color=#444444]液相色谱做定量时,如果标准品和样品称量一样,进样量也一样,是不是说,物质的量之比就等于峰面积之比呢?[/color]
关于液相色谱的定量分析定量分析是在定性分析的基础上,需要纯物质作为标准样品。液相色谱的定量是相对的定量方法,即:由已知的标准样品推算出被测样品的量。
岛津的液相色谱,配有自动进样器。用外标定量时,只配一个标样,选择用改变的进样量(即1微升,2微升。。。。)这种方式来做标准曲线,是不是线性更好一些。
我用自动进样,定量环为100微升的液相色谱分析样品,现进样量要改为10微升,进样能准确吗?
各位前辈,请教一个问题,关于液相色谱定量时对于有检出的阳性样品复测是否一定要走标液曲线,如果用单点定量的检测值的准确度是否会与标液曲线定量的值相差较大;对于阳性样检出值接近检测限的情况两者哪个更有优势,毕竟走单点可以节省很多时间。。。。
[color=#444444]最近在用液相色谱仪做水杨酸的定量,水杨酸用甲醇:水=60:40配制,流动相为甲醇:水(0.01M甲酸铵)=60:40,但是不知道什么原因,水杨酸的面积随着时间的推移一直在变小?另外,问下大家是用什么方法做的[/color]
[color=#00008B][B]该帖为楼主自己整理,帖中所有楼主撰写的内容未经授权不得转载,否则属侵权违法行为![/B][/color]看到很多版友发帖求助讨论准确性的问题,我特意整理了一个关于定量分析误差问题的帖子,希望对大家有所帮助。高效液相色谱定量分析过程可分为样品的前处理、标准品的配制、进样、色谱分离、检测及数据处理等七个步骤。[B]一 误差的主要来源[/B]随着现在市场销售仪器自动化程度的提高,进样、色谱分离、检测及数据处理等实验环节对实验结果产生的误差越来越小,尤其在高效液相色谱定量分析中,实验结果的误差可能主要来源于样品的前处理及标准品的配制。[B]1、样品的前处理 [/B]样品的萃取率是样品前处理时存在的主要问题。当固-液萃取(含柱分离的前处理方法),液-液萃取时,存在萃取率不高且不稳定的问题。尤其在除去蛋白质时,存在变性蛋白质会吸附一些被测组分,导致萃取率降低的情况。通常,萃取率是通过在式样中添加被测成分在萃取的方法评价的。也就是说,被测成分的增加量和液相色谱中的峰面增大成比例关系,通过这种方法可以确定溶液中被测成分的变化趋势。 如果存在萃取率不稳定的问题,就有必要改变萃取的方法,要预先添加内标,然后萃取。这种情况采用的内标,必须与被测物质的化学结构类似,萃取的萃取率才可能相近。如果回收率不但接近100%而且较为稳定,可以证明这种前处理方法较为可靠。在分析中如果能充分考虑以上误差产生的各种原因,才有可能得到精确的分析结果。 [B]2、标准品的配制[/B]本帖中讨论的问题带有普遍性,影响高效液相色谱分析结果准确性的因素较多,仅在标准溶液的配置过程中,可以分为标准物质的称量,溶液的配制和溶液的储存三个环节。 作为标准溶液使用的标准物质其纯度要求很高,应避免使用纯度不符合要求的试剂,作为标准溶液使用的标准物质具有不可替代性,现在市场有销售的HPLC专用的溶剂及各种标准试剂可供实验选择;其次选择与样品浓度要求相适应的天平,应该尽可能使用高精度的天平,这样才能把由于天平使用带来的称量操作误差降至最低。[B]二 消除误差的方法[/B] 要提高分析结果的准确度,必须考虑在分析过程中可能产生的各种误差,采取有效措施,将这些误差减到最小。[B]1、选择合适的分析方法[/B] 各种分析方法的准确度是不同的。化学分析法对高含量组分的测定能获得准确和较满意的结果,相对误差一般在千分之几。而对低含量组分的测定,化学分析法就达不到这个要求。仪器分析法虽然误差较大,但是由于灵敏度高,可以测出低含量组分。在选择分析方法时,一定要根据组分含量及对准确度的要求,在可能条件下选最佳分析方法。[B]2、增加平行测定的次数[/B] 如前所述增加测定次数可以减少随机误差。在一般分析工作中,测定次数为2—4次。如果没有意外误差发生,基本上可以得到比较准确的分析结果。[B]3、消除测定中的系统误差[/B] 消除测定中系统误差可采取以下措施:其一是做空白实验,即在不加试样的情况下,按试样分析规程在同样操作条件下进行的分析。所得结果的数值称为空白值。然后从试样结果中扣除空白值就得到比较可靠的分析结果。其二是注意仪器校正,具有准确体积的和质量的仪器,如滴定管、移液管、容量瓶和分析天平,都应进行校正,以消除仪器不准所引起的系统误差。因为这些测量数据都是参加分析结果计算的。其三是作对照试验,对照试验就是用同样的分析方法在同样的条件下,用标样代替试样进行的平行测定。将对照试验的测定结果与标样的已知含量相比,其比值称为校正系数。 校正系数=标准试样组分的标准含量/标准试样测定的含量 被测试样的组分含量=测得含量×校正系数 综上所述,在分析过程中检查有无系统误差存在,作对照试验是最有效的办法。通过对照试验可以校正测试结果,消除系统误差。[B]4、样品定量分析过程中的误差[/B]样品处理要尽量减少操作者的技术问题带来的误差,样品的稀释次数、稀释工具都是误差的祸根,应尽量减少稀释次数,稀释工具用高准确度的。样品中的干扰组分会直接影响分析的准确度,而且有些组分会损坏柱子。纯化样品的过程尽量少用蒸发至干的步骤(在色谱分析中这一步又是不可少的),正确操作固相柱萃取、纯化小柱使用的步骤,注意提高每一步的回收率,使用内标法也是一个能准确定量的方法。 在手动进样中进样体积至少是样品定量环管体积的3倍,色谱分离程序要使色谱峰的分离度大于1.5,控制流动相、流量、温度等的平稳。流动相的污染都会抬高基线或减少信噪比,分辨率下降,试验条件的变化,如柱退化、不好的流动相等都能引起保留时间变化,引起一个峰或更多的峰不能被鉴别。正确设定仪器参数,选用合理的数据处理参数,用峰面积计算结果比峰高更精确。[B]三 结论 [/B]以上的分析的是我们能尽量控制的误差,还有一些不是操作者所能控制的误差,如被测定组分易分解、组分的含量高低、介质效应等。我们把能控制的误差减小到最低,那你的结果准确度将更高。
我用自动进样,定量环为200微升的[url=https://insevent.instrument.com.cn/t/5p][color=#3333ff]液相色谱[/color][/url]分析样品,现进样量是5微升,进样能准确吗? 会不会导致对照品连续进样5针,峰面积忽大忽小?
液相色谱如何进行定性和定量分析?
请问许多文献上提出核磁内标法(绝对测量法)定量分析时不需引进任何校正因子,不需制作标准曲线,但高效液相色谱为何需制作标准曲线?高效液相色谱和核磁内标法定量的原理差不多呀?[em09]
高效液相色谱串联质谱可以定量吗?
液相色谱法定量用外标法定量时,含量超过100%的可能性有多少,可信吗?有多少改善的余地?
各位,液相色谱定量优于气质,具体原因要如何解释?给人培训,要比较专业的术语来解释,一下把我难到了
液相色谱分析间苯二磺酸和苯磺酸的条件及定量方法?
前几天做的实验能用液相色谱能把饱和分、芳香分和胶质分开,但只是知道各部分出的峰的面积,因为饱和分、芳香分和胶质都不是纯物质,而且不同物质的同一组分性质又不一定相同,所以无法知道每一组分的校正因子,不知道如何定量,请高手帮忙!
用液相色谱测定化合物的含量时,没有标准品,目前只能用面积归一化来定量,我感觉这样很不准确,大家有什么好的办法没有?
硼砂做液相色谱,外标定量,峰面积相差不大,
液质联用仪有国家标准,但没有液相色谱仪的检测标准,我能参考液质的前处理和液相色谱仪的条件,用液相色谱仪检测吗?
假如:测有机酸的柱,液相色谱柱与[url=https://insevent.instrument.com.cn/t/3p][color=#3333ff]离子色谱[/color][/url]柱能通用?我是初学着,资金有现,不要笑话我?
液相色谱分离条件能模拟吗?
液相色谱柱能串联和并联使用吗?
[color=#444444]液相色谱进行简单的峰面积比定量分析时,如果合成的样品的溶剂是四氢呋喃,而标准品的溶剂是甲醇,而且合成的样品产量很少,请问如何将四氢呋喃转换成甲醇?是否可以通过氮气吹干四氢呋喃溶剂;是否还有别的可行的方法?[/color]
各位大侠; 我现在使用的液相色谱定量方法是在建立方法时坐标准曲线,在以后测定样品含量时就一直套用以前的标准曲线求值,现在问题是怎么确保曲线的可用性,系统的稳定性一直好可以一直用以前的曲线求值吗?
液相色谱和[url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]的外标法和内标定量有何区别?是如何处理的?
各位大神,实在木有办法,对这一片太不熟悉了,能帮我看看这道题吗?一台高效液相色谱进样环为20ul,做定量分析,一次进样为26ul,另一次进样为18ul,这两次进样对分析结果分别有什么影响
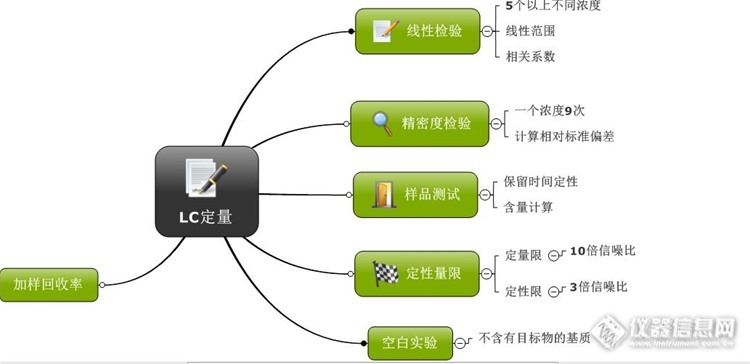
今天读到傅若农教授的文字,感到很温暖。想想我这个分析化学战线的逃兵,将彻底地和分析化学说再见了。但是,我还是讲我的一些思路做一个总结,希望能给我们90后的分析工作者有点启发。接着去年我那个帖子。《谈谈液相色谱方法开发》,有网友希望我写点定量方面的内容。那么,今天,我权当抛砖引玉。当然,我写的很不成熟,希望大家批评指正。http://bbs.instrument.com.cn/topic/5957822在做好了分离方法之后,我们需要对我们的目标物进行定量分析。定量分析的思路一般是http://ng1.17img.cn/bbsfiles/images/2016/06/201606241414_597987_1626663_3.png一般来说,通过这套思路来进行定量方法学的验证。这个是一个通用的方法。当然,这也是十五多年前我做课题时候。 我导师教我的,现在也许也有发展了吧。
做液相色谱的童鞋们,哪些故障你能自己处理?
[url=https://insevent.instrument.com.cn/t/Mp][color=#3333ff]气相色谱[/color][/url]柱和[url=https://insevent.instrument.com.cn/t/5p][color=#3333ff]液相色谱[/color][/url]柱能通用吗?
跟大家分享一下液相色谱讲义,里面包括(1)液相色谱基础知识(2)液相色谱的方法开发-分离机理及色谱柱(3)分离方法,离子抑制及离子对方法介绍(4)梯度方法的开发及梯度分离时的注意事项(5)液相色谱的实用技术,色谱柱的保养、样品前处理(6)液相色谱的定性、定量分析(7)色谱泵和进样器的原理及操作(8)检测器的原理及操作
我是做本科毕业设计的,用茴香脑制备茴香醛。 产品分析用的是液相色谱,现在需要做一条原料的标准曲线,但是原料的出峰位置不固定,不仅不同浓度的原料溶液出峰位置不一样,相同浓度进针走样出峰位置也会变。还有一个问题是:峰很宽,跨度有2-3分钟。 溶质:茴香脑,溶剂:甲醇。 液相色谱流动相:水:甲醇:乙腈=5:3:2。 怀疑溶液和流动相发生了反应,所以配了另一瓶茴香脑甲醇溶液,加入了一定量乙腈,尝试用气相色谱分析,定性地看看出峰位置是否一样(因为气相色谱不能用水,所以只加入了乙腈),结果是出峰位置都在9-10分钟之间。 是否有高手能告诉我问题出在哪里,以及怎么解决?PS:液相色谱做其它分析都正常的,学姐在做的茴香醚制备茴香醛也没问题。







 我要推广仪器
我要推广仪器
 下载APP
下载APP