我们用的是安捷伦1100的高效液相色谱仪,在做一个软膏剂的时候,标准规定的浓度进样10μl,峰形有前延峰,把浓度稀释一半,进样10μl,峰的对称性在1.0左右,如果再把浓度稀释一半,进样20μl的话,峰形又不对称了。大家分析一下是什么原因?液相色谱的进样量与峰形之间的关系怎样?(药品名字是“复方地塞米松乳膏”,内标法测定地塞米松含量,内标物是甲睾酮)
请大家谈谈液相色谱中样品溶液的浓度问题,已同事说可以看色谱图中的峰面积,差不多在7000000左右,还有就是响应值在500mV左右,他说的有道理吗?还是必须要做浓度线性关系呢?
做高效液相色谱标准曲线时,将标准品配制成一个浓度,然后将该样品按照1uL,3uL,5uL,7uL,9uL的进样量进行进样,然后做成标准曲线,相关系数为0.99998。这样做标曲可以么?在做未知含量样品时应该按照多少的进样量进行进样呢?请高人指教,不胜感激!
制备液相色谱的上样量与什么有关
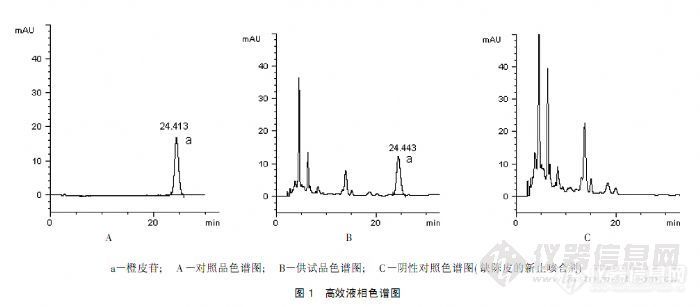
作者:李仁秋 (云南省昆明市儿童医院, 云南 昆明 650011)摘 要:目的: 建立高效液相色谱法测定新止咳合剂中橙皮苷的含量。方法: 采用 Dikma 钻石 C18 色谱柱( 4. 6mm @250mm, 5Lm) ; 流动相为甲醇) 0. 2% 磷酸溶液( 38: 62) ; 检测波长为 283nm,流速为 1. 0ml/ min, 柱温为 30 e 。结果: 橙皮苷进样量在 0. 01~ 2. 0Lg ( r= 0. 999 97)范围内与峰面积线性关系良好,平均加样回收率为 99. 12%, RSD= 0. 84%( n= 9) 。结论:本法简便、 快速、 专属性强、 重现性好;可作为新止咳合剂的质量控制方法。关键词:橙皮苷; 高效液相色谱法;新止咳合剂; 含量测定http://ng1.17img.cn/bbsfiles/images/2012/07/201207161641_377910_2379123_3.jpg
本人刚接触液相色谱,想要检测某饮料中某一物质的含量采用外标法,该物质的大体含量不清楚,看了几篇参考文献说外标法中浓度范围最好包括样品浓度,这该怎么做呢?难道扩大外标法中的浓度范围吗,这样好像比较麻烦又不见得准确啊?还有就是进样量应该怎么确定啊?
液相色谱柱在装填时,是从一个方面装填的,那两头是完全一样的吗?
高效液相色谱进样量一般为多少啊
高效液相色谱进样量对分析结果有怎样的影响?
用[url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]和液相色谱如何测定污水的化学耗氧量和生物需氧量?谢谢!

最近,经常看到有客户询问我们液相色谱柱的最大上样量是多少?其实这个问题不好回答,因为最大上样量与很多因素有关,小版今天翻阅迪马综合目录,大有收获,或许可以简单说明一下这个问题下图贴出来与大家共享http://ng1.17img.cn/bbsfiles/images/2015/05/201505211648_546950_1610895_3.jpg
液相色谱一般都有定量环 20ul的居多 但是我们进样有时候进20有时候进10的 进20ul时候、大家一般用20ul定量环时候进多少 有人说 进三倍量 那如果近10ul的是不还要换定量环啊 我一直都是20的 从没有换过 一般也没有按照进3倍量!
[color=#444444]请问,液相色谱测定含量双样双针的,f值有没有统一规定的啊?F=称样量/A峰面积/稀释倍数,要不要除以对照品的稀释体积啊?是随便写还是有统一规定写法啊?[/color]
制备液相色谱的上样量与什么有关
用液相色谱检测人血白蛋白中多聚体含量时,乙酰色氨酸的峰走成了两个峰,试验用的是凝胶柱,用的PH7.0的磷酸盐缓冲液,请老师们指教下,这是怎么回事,柱子是新的,才用几天,之前都是正常的,昨天用纯水冲洗柱子两个小时!今天进同样的样品,走出来的峰是一样的下图,一个是正常的,一个是走成两个峰的,请老师们指教http://ng1.17img.cn/bbsfiles/images/2014/03/201403071204_492237_2623284_3.jpghttp://ng1.17img.cn/bbsfiles/images/2014/03/201403071205_492239_2623284_3.jpg
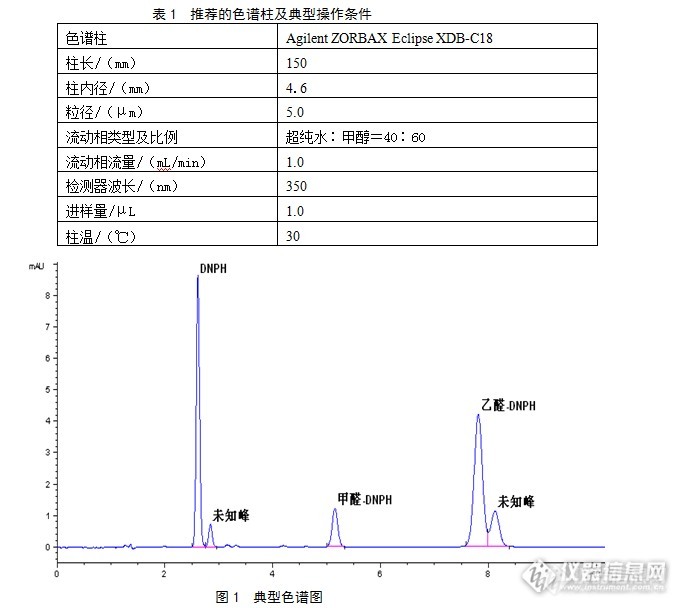
微量甲醛、乙醛液相色谱测定法1 适用范围本标准规定了液相色谱测定水溶液或环氧乙烷样品中的甲醛和乙醛的方法。本标准适用于样品中甲醛、乙醛含量为1~50mg/kg的分析。2 方法概要 液态样品中微量甲醛、乙醛与2、4-二硝基苯肼衍生生成甲醛-2、4-二硝基苯腙(以下称:甲醛-DNPH)和乙醛-2、4-二硝基苯腙(以下称:乙醛-DNPH),在通过固相C18小柱将甲醛、乙醛的衍生物从溶液中萃取出来。随后,使用自动进样器直接将萃取出的甲醛、乙醛衍生物注入[url=https://insevent.instrument.com.cn/t/Mp]气相色谱仪[/url],样品在甲醇和水混合溶液携带下通过毛细管色谱柱分离,使样品里的甲醛衍生物、乙醛衍生物及未反应2、4-二硝基苯肼按照不同顺序先后进入紫外检测器检测。最后,由色谱数据工作站采用外标法得到各物质含量。3 试剂和材料3.1 甲醇:液相色谱(HPLC)纯。3.2超纯水:比电阻率大于18.2ΜΩcm的水。3.32,4-二硝基苯肼(DNPH):分析纯。3.4超声波振荡器。3.5玻璃溶剂过滤器。3.6真空泵。3.7量筒:10mL。3.8量筒:25mL。3.9具塞碘量瓶:250mL。3.10C18固相萃取小柱(SPE)。3.11注射器:5mL。3.12注射器:10mL。3.13烧杯:50mL3.14标准溶液:与分析样品浓度相接近甲醛、乙醛水溶液,自配或外购。4 仪器4.1液相色谱仪:安捷伦科技公司1200型,配有自动进样器、真空脱气、四元梯度泵和紫外检测器,或同类产品。4.2色谱数据工作站:安捷伦科技公司Chemstation工作站,或同类产品。4.3色谱柱:见表1推荐色谱柱,或同类产品。推荐色谱柱典型操作条件见表1,典型色谱图见图1。[img=,681,608]http://ng1.17img.cn/bbsfiles/images/2017/09/201709032112_01_2166779_3.png[/img]5 校正仪器第一次使用或者被改变了某一或某些参数设置(如流量、流动想类型、流动相比例、色谱柱等),应对仪器进行校正。使用已知与样品接近含量的标准甲醛、乙醛溶液与DNPH络合后,在液相色谱仪上分析,测量两物质色谱峰峰面积,计算校正因子k[sub]i[/sub]:k[sub]i[/sub]=标准液中i物质浓度/i物质衍生物峰面积。6 试验步骤6.1用5mL注射器将5mL甲醇注入C18固相萃取小柱,冲洗萃取小柱进行预处理,然后再用5mL超纯水冲洗。6.2分别取10g样品(由于环氧乙烷沸点较低,环氧乙烷产品可直接使用量筒量取11.2mL)、10mL2,4-二硝基苯肼(DNPH)试剂和10mL超纯水转移到碘量瓶内,盖上瓶塞,让混合物反应15分钟。如果观察到沉淀,则应使用超纯水按1:10稀释样品,并按上述描述再次进行操作。6.3使用一个10mL量筒量取10mL的反应混合物,用针筒将其转移到一个6.1中预处理好的固相萃取小柱,注射时应将样品缓慢地推进萃取小柱。然后,使用10mL超纯水重复上述步骤,注意需将所有液体推出萃取小柱。6.4用注射器将8mL甲醇推进萃取小柱,以洗脱富集被固相萃取小柱从样品中萃取出的甲醛-DNPH、乙醛-DNPH及未反应的DNPH溶液。洗脱富集液放置于烧杯中。再向烧杯添加少量甲醇,使溶液的最终重量达到10±0.01g。6.5使用符合4.3中推荐色谱柱或同类色谱柱,采用自动进样器将适量6.4中的洗脱富集样品注入4.1规定液相色谱仪,通过色谱工作站测量各物质出峰面积,利用已测校正因子面积归一定量计算样品中各组成含量。6.6用10mL的去离子水替代样品重复步骤6.2至6.5,测定空白。7 计算7.1溶液甲醛、乙醛含量按下式计算:(外标法)X[sub]i[/sub]=A[sub]i[/sub]×k[sub]i[/sub]式中:X[sub]i[/sub]——被测组分i的含量,mg/kg;A[sub]i[/sub]——被测组分i的衍生物峰面积;k[sub]i[/sub]——被测组分i的绝对校正因子;i——甲醛或乙醛。8 精密度8.1 重复性同一操作者重复测定的两次测定结果的差值应不超过平均值的10%(m/m)。9 结果的表示仪器稳定状态下连续两次测定结果的平均值作为出厂产品的测定结果,生产装置过程控制使用单次测定数据作为结果报告,结果的表示修约至1mg/kg。10注意事项10.1 仪器进样前取样,需摇晃采样容器,以保证分析样品的均匀性。10.2 色谱流动相必须使用液相色谱(HPLC)纯试剂。10.3当分析环氧乙烷或含有环氧乙烷样品时,使用前应事先将试剂、容量瓶和量筒冷却,以防止环氧乙烷受热挥发。10.4 对含有环氧乙烷的样品的任何操作,必须在通风柜中完成,并做好个人防护。
液相色谱进样量一样出峰为什么不一样
[align=center][font='calibri'][size=13px][url=https://insevent.instrument.com.cn/t/5p][color=#3333ff]液相色谱[/color][/url]分析中[/size][/font][font='calibri'][size=13px]进样量对[/size][/font][font='calibri'][size=13px]测定结果的影响[/size][/font][/align][align=center][font='calibri'][size=13px]通标小菜[/size][/font][font='calibri'][size=13px]鸟[/size][/font][/align][font='calibri'][size=13px]在[url=https://insevent.instrument.com.cn/t/5p][color=#3333ff]液相色谱[/color][/url]分析中,样品[/size][/font][font='calibri'][size=13px]进样量的[/size][/font][font='calibri'][size=13px]选择直接关系到我们色谱出峰的好坏,一旦[/size][/font][font='calibri'][size=13px]进样量过高[/size][/font][font='calibri'][size=13px],[/size][/font][font='calibri'][size=13px]出峰就[/size][/font][font='calibri'][size=13px]会异常,进而会导致目标物组分无法准确定量和定性分析。正常[/size][/font][font='calibri'][size=13px]的出峰色谱图[/size][/font][font='calibri'][size=13px]是一种对称的,峰宽合适,峰顶部尖锐的符合正态分布的图形,如下所示:[/size][/font][align=center][img]https://ng1.17img.cn/bbsfiles/images/2023/07/202307011749508632_3015_3141805_3.jpeg[/img][/align][font='calibri'][size=13px][url=https://insevent.instrument.com.cn/t/5p][color=#3333ff]液相色谱[/color][/url]分析中[/size][/font][font='calibri'][size=13px]进样量对[/size][/font][font='calibri'][size=13px]色谱出峰的影响主要分为以下两类:[/size][/font][font='calibri'][size=13px]1.进样体积超载[/size][/font][font='calibri'][size=13px]在日常分析样品过程中,对于一个未知浓度的样品,我们往往无法准确估计其浓度大小,因而为了保险起见,都会进行大体积进样,确保其在色谱上有响应,能够正常出峰。然后对于浓度较小的试样来说,大体积进样对出峰影响不大,但是对于浓度较高的样品,仅仅只是进样10ul,甚至是5ul,色谱出峰就会异常,如下图所示:[/size][/font][align=center][img]https://ng1.17img.cn/bbsfiles/images/2023/07/202307011749511182_2329_3141805_3.jpeg[/img][/align][font='calibri'][size=13px]我们可以从图中清楚的看到色谱峰的峰宽正常,峰高也正常,但在峰顶[/size][/font][font='calibri'][size=13px]点除处会[/size][/font][font='calibri'][size=13px]出现一个平台,我们常常将其称之为“平头峰”。[/size][/font][font='calibri'][size=13px]其实这是[url=https://insevent.instrument.com.cn/t/5p][color=#3333ff]液相色谱[/color][/url][/size][/font][font='calibri'][size=13px]进样量过大[/size][/font][font='calibri'][size=13px]导致信号过大,信号超过记录仪的最大测量值,不再上升而出现平头峰。遇到这种情况我们应该从以下几个方面来解决:[/size][/font][font='calibri'][size=13px]a.减少进样体积,[url=https://insevent.instrument.com.cn/t/5p][color=#3333ff]液相[/color][/url]不像[url=https://insevent.instrument.com.cn/t/Mp][color=#3333ff]气相[/color][/url]可以通过调整分流比来[/size][/font][font='calibri'][size=13px]减小进[/size][/font][font='calibri'][size=13px]样量,[url=https://insevent.instrument.com.cn/t/5p][color=#3333ff]液相[/color][/url]进[/size][/font][font='calibri'][size=13px]样通过[/size][/font][font='calibri'][size=13px]定量环进样,因而可以在软件中设置小的进样体积,比如设置1ul、2ul的进样体积;[/size][/font][font='calibri'][size=13px]b.适当调节检测器信号衰减,改变记录仪量程;[/size][/font][font='calibri'][size=13px]c. 增大色谱仪上衰减倍数,减小灵敏度;[/size][/font][font='calibri'][size=13px]2.试样质量超载[/size][/font][font='calibri'][size=13px]试样质量超载也是过载的一种,尽快我们选择的进样体积很小,但也有可能因为试样中目标物组分的质量太高或者说浓度太高,同样也会造成柱过载,导致[/size][/font][font='calibri'][size=13px]样品峰展变宽[/size][/font][font='calibri'][size=13px],峰型改变,保留时间漂移。[/size][/font][align=center][img]https://ng1.17img.cn/bbsfiles/images/2023/07/202307011749510566_4189_3141805_3.jpeg[/img][/align][font='calibri'][size=13px]这种情况下的色谱出[/size][/font][font='calibri'][size=13px]峰一般[/size][/font][font='calibri'][size=13px]为前倾峰,随着样品浓度的增加,峰会前倾的越来越严重,直至变成一个类似于[/size][/font][font='calibri'][size=13px]直角三角形得峰型[/size][/font][font='calibri'][size=13px]。[/size][/font][font='calibri'][size=13px]以上两种[/size][/font][font='calibri'][size=13px]出峰情况[/size][/font][font='calibri'][size=13px]都是不正常的出峰(鉴定杂质故意使其过载除外),如果在实际分析中遇到这两种情况,我们只需要对样品进行稀释减小浓度或者[/size][/font][font='calibri'][size=13px]减小进[/size][/font][font='calibri'][size=13px]样体积就能避免类似情况的发生。[/size][/font][align=center][font='calibri'][size=13px] [/size][/font][/align]
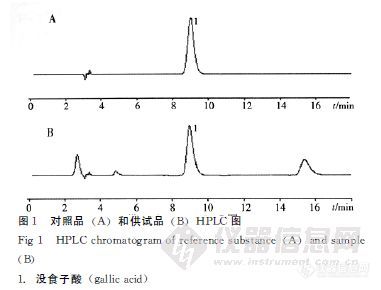
【作者中文名】 刘英慧; 陈晓玲; 黄琪; 雷鹏; 梁敏红; 【作者单位】 中南大学湘雅医院药剂科; 中南大学药学院; 江苏省军区机关医院; 【摘要】 目的建立高效液相色谱法测定檵木叶中没食子酸的含量。方法以Diamonsil C18(4.6mm×250mm,5μm)色谱柱为分析柱;甲醇-水-0.1%磷酸(12∶78∶10)为流动相;流速为1.0mL.min-1;检测波长为273nm;柱温:30℃;进样量10μL。结果没食子酸进样量在0.039 8~0.498 0μg呈现良好的线性关系(r=0.999 8),平均回收率为99.7%,RSD为2.7%。结论本法操作简便、准确,具有良好的重现性,可用于檵木叶中没食子酸的含量测定,为檵木叶药材的合理应用及质量控制奠定了基础。http://ng1.17img.cn/bbsfiles/images/2012/08/201208011237_381012_1761902_3.jpg
当用[url=https://insevent.instrument.com.cn/t/5p][color=#3333ff]液相色谱[/color][/url]溶剂极性强度大的试剂,如纯甲醇、纯乙腈,纯乙醇,而分析体系中以水为主时,如果样品进样量大,如定量管为20ul,此时单一的纯物质会出现双峰,第二峰比第一峰小(每次都不太一样),且拖尾,保留时间会提前(相对进样量少而言),将进样量减少一半以上,峰型将变为正常。这是样品的溶剂与流动相极性相差太大,而流动相来不及将其稀释达到平衡造成的,此情况下需要减少进样量。另一个原因是,进样量不一定大,但绝对量很大,色谱图上的双峰紧靠在一起,基本上齐高,不拖尾(如果出峰很快,也可能是色谱柱问题)。将样品稀释再进样就可以了,这是由于进样量过大,色谱柱过载造成的。

摘要: 目的 建立健胃丸中橙皮苷的含量测定方法。方法 液相色谱法。色谱柱:Kromasil C18柱(4.6mm×250mm,5µm);流动相:甲醇-0.5%冰醋酸溶液(35:65);流速1.0ml/min;柱温:40℃;检测波长:280nm。结果: 橙皮苷在0.2403~1.1214μg 范围内呈线性关系(r=0.9998,n=5),平均加样回收率为99.24 % RSD为0.35 % (n=6)。结论 该方法操作简便,结果可靠,可用于健胃丸中橙皮苷的含量测定。关键词:高效液相色谱法;健胃丸;橙皮苷。健胃丸主要有陈皮、山楂、神曲B6等成分组成的复方制剂 ,用于消化不良和饮食阻滞等辅助治疗。为了有效控制健胃丸的质量,经过资料检索以及反复实验探索,本文运用高效液相色谱法对其中主要成分橙皮苷进行了定性定量的研究,建立了健胃丸的含量测定的高效液相色谱法。样品处理简单,结果理想,可以作为健胃丸质量控制方法。1 仪器与试药1.1 仪器岛津LC-20AT高效液相色谱仪,SPD-M20A检测器, LCsolution色谱工作站, Metteler AB265S(0.01mg);Sartorius BS124S(0.1mg)1.2 试药 橙皮苷对照品(中国药品生物制品检定所,批号110721-201014);甲醇为色谱纯,冰醋酸为分析纯,水为纯化水。健胃丸(医药制剂自制,批号:20120531、 20120624、20120705 ,规格为0.3g)2 实验方法与结果2.1 色谱条件与图谱色谱柱: Kromasil C18柱(4.6mmx250mm,5um);检测波长:283nm;流速:1.0ml.min-1;流动相:甲醇-0.5%冰醋酸 (35:65);柱温:40℃。橙皮苷在14min左右出峰,阴性无干扰。图谱如下:http://ng1.17img.cn/bbsfiles/images/2012/10/201210311902_400538_1839779_3.jpghttp://ng1.17img.cn/bbsfiles/images/2012/10/201210311903_400539_1839779_3.jpg2.2 溶液制备2.2.1对照品溶液 精密称取橙皮苷对照品6.74mg,置于50ml量瓶中,加甲醇溶解并稀释至刻度,摇匀,精密量取6ml置50ml量瓶中加甲醇稀释至刻度,摇匀,作为对照品储备液。2.2.2供试品溶液取本品内容物混匀,,取约0.15g,精密称定,置具塞锥形瓶中, 精密加入甲醇10ml,称定重量,超声50分钟使溶解 ,放冷,再称定重量,,用甲醇补足减失的重量,摇匀,用0.45μm的微孔滤膜滤过,滤液作为供试品溶液。2.3 线性关系考察精密量取对照品储备液5、10、15、20、25μl,按上述色谱条件,记录色谱图,测定峰面积。以橙皮苷面积为纵坐标,橙皮苷进样量为横坐标(ug),得回归方程:Y=184537.97X+4155.6,相关系数 r=0.9998(n=5) ,结果表明橙皮苷在0.2403~1.1214μg[/fo
甜菜碱、甜菜碱盐酸盐产品怎样用高效液相色谱分析?谁有分析方法?产品是甜菜碱(三甲胺乙内酯)、甜菜碱盐酸盐,还有维生素(A\B\C\D\E),这几样产品怎么用液相色谱分析纯度(含量)?维生素系列能不能用紫外检测器全做了?
据权威统计,当前国内一年的[url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]需求量为6000台,一半为进口的;液相色谱为5000台,9成为进口的。各位老大对此数据有何感想?
高效液相色谱测定明胶分子量的方法??要选用什么样的流动相、缓冲溶液、还有标准曲线用什么样的标准物质??可以的话最好有具体的步骤
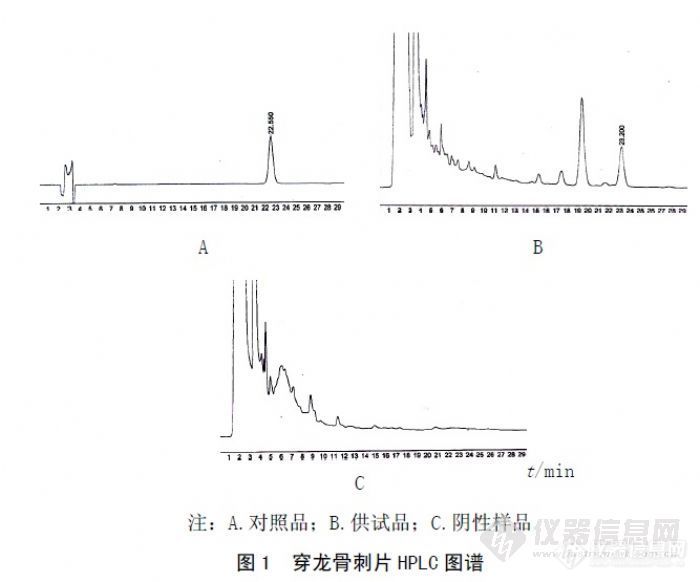
作者:丁永志;李霄锋;蔡俊安;郭鑫慧;(河南百年康鑫药业有限公司;河南财鑫集团有限责任公司;)摘要:目的采用高效液相色谱法测定穿龙骨刺片中淫羊藿苷的含量。方法采用外标一点法,Diamonsil ODS1 C18色谱柱,乙腈-水(27∶73)为流动相,流速为1.0mL/min,检测波长270nm。结果淫羊藿苷在0.125~1.00μg范围内呈良好线性,回归方程为Y=81601X+10356,r2=0.9997,平均加样回收率为99.12%,RSD=0.65%(n=6)。结论本方法简便、准确,专属性强,测定结果重复性好,为穿龙骨刺片中淫羊藿苷的定量分析提供了科学有效的方法。谱图:http://ng1.17img.cn/bbsfiles/images/2012/08/201208131051_383402_1606903_3.jpg
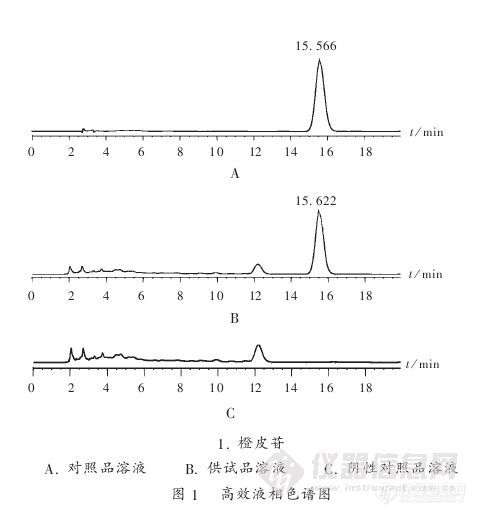
【作者】 史大军; 袁才琼; 邱海蕴;【Author】 Shi Dajun,Yuan Caiqiong,Qiu Haiyun(Yichang Municipal Institute for Food and Drug Control,Yichang,Hubei,China 443005)【机构】 湖北省宜昌市食品药品监督检验所;【摘要】 目的建立测定小儿吐泻宁中橙皮苷含量的高效液相色谱(HPLC)法。方法以Diamonsil C18柱(250 mm×4.6 mm,5μm)为色谱柱,甲醇-冰醋酸-水(35∶4∶61)为流动相,流速1.0 mL/min,检测波长283 nm,采用外标法定量。结果橙皮苷进样质量浓度线性范围为3.58~143.2μg/mL(r=0.999 9);平均回收率为98.96%,RSD为0.80%(n=6)。结论该方法简便、快速、准确,适用于小儿吐泻宁的质量控制。 http://ng1.17img.cn/bbsfiles/images/2012/08/201208201700_384763_2379123_3.jpg
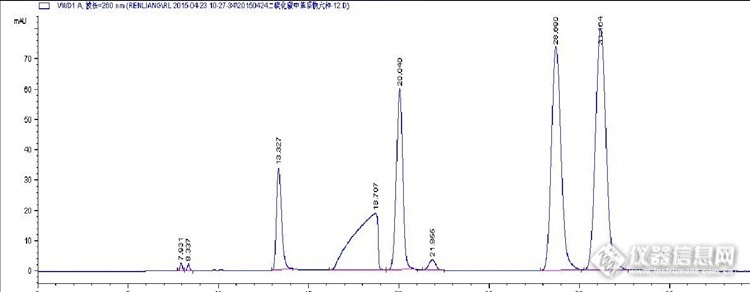
近些年来,随着科技的飞速发展,液相色谱技术应用也逐渐步入快车道,沃特世、岛津、安捷伦、戴安等分析仪器知名公司都相继研发成功并将最新的的超高效液相色谱仪推向市场,业界内外纷纷一味追求高精尖,追逐快速高效,殊不知液相色谱类仪器并非一味追求快速,而是好比谈恋爱,要看是不是适合自己的工作要求,这其中难免就会涉及到分析项目、分析精度以及工作成本等等一系列因素,本文就自己心得以及所学在此做与大家一起来探讨交流: 首先,众所周知,高效液相色谱是以液体为流动相,采用高压输液系统,将具有异极性的单一溶剂或不同比例的混合溶剂、缓冲液等流动相泵入装有固定相的色谱柱,在柱内各成分被分离后,进入不同类型的检测器进行检测,从而实现对样品的定性定量分析。而超高效液相色谱与高效液相色谱原理基本相同,不同之处就是:1、色谱柱发生了变化 小颗粒高性能微粒固定相出现,2、流动相超高压输液泵出现 3、采集速度更快的检测器出现 ,主要因素就是基于这三点。但是,超高效液相色谱仪并非针对每一种分析样品,每一种分析化合物都如此高效快速,必须因样而选,否则的话,就会适得其反,事倍功半,前功尽弃。http://ng1.17img.cn/bbsfiles/images/2015/08/201508201023_561650_2328678_3.jpg 笔者单位去年采购一台 Waters ACQUITY UPLC H-CLASS,在国内环境监测业界还算是高大尚的液相色谱仪器,根据介绍可以实现:无需更改现有工作流程即可获得UPLC性能灵活和简便的四元溶剂混合和直接注射取样 完美兼容HPLC方法,无缝转换至UPLC方法 方法转换包可保证分离方法的一致性,流速范围0.010 ~ 2.000 mL/min,以0.001 mL增速步进,最高操作压力:15,000 psi最高到1 uL/min,9,000 psi最高到2 mL/min,据于此类性能,加之单位以前有一台服役多年面临脱保的安捷伦LC-1100,可以接班服役,同时提高工作效率,不必再劳神费心,一个样品等待大把时间,因此,趁着还在维保范围内,迅速入手,希望短时间之内形成"作战“能力,投入实战。 恰逢迪马科技在论坛搞一个色谱柱试用活动,遂参加尝试用液相色谱法分析水中苯系物,以做直观比较:色谱柱型号:迪马 DiamonsilPlus (固定相),250mm(柱长)4.6 mmx5 μm色谱柱型号:Agilent Eclipse PAH固定相),250mm(柱长) 4.6 mm x 5 μm WATERS XBridge C18 250mm(柱长)4.6 mmx 5 μm 以及 UHPLC WATERS 50mm(柱长)2.1mm x 1.7 μm,其中HPLC分析条件如下: 流动相: 甲醇 :水= 65: 35 (如有梯度请注明梯度表)流速: 1.0 mL/min柱温: 40 ℃检测器:UV 260 nm 或其他检测器 进样量: 10 μL分析结果如下:Diamonsil Plus谱图http://ng1.17img.cn/bbsfiles/images/2015/08/201508201112_561654_2328678_3.jpgAgilent Eclipse PAHhttp://ng1.17img.cn/bbsfiles/images/2015/08/201508201113_561655_2328678_3.jpgWATERSXBridge C18http://ng1.17img.cn/bbsfiles/images/2015/08/201508201114_561656_2328678_3.jpg上述结果分析:分析比较:Diamonsil Plus与Agilent EclipsePAH WATERS XBridge C18所分析谱图比较:前者为甲醇中的六中苯系物谱图,后者为二硫化碳中的苯系物,其他条件相同,前者流速为1mL/min进样量为20ul,后者流速为0.5mL/min进样量为5ul,通过比较可以基本得出如下结论:Diamonsil Plus 可以承受大进样量的化合物分析,柱效表现良好,在20/5的情况下,峰形保持较好;Agile
液相色谱,面积归一化进样量与峰面积有关系吗?
液相色谱检测(被测物是纯度比较高的)制成一定系列浓度,再用液相色谱检测,是不是进样浓度越高,峰值就越大
溶液是用蒸馏水配置的,进样前先过滤,过滤后是否可以直接进水样进行分析呢?除了过滤还有什么要注意的吗?如果同样的浓度,但是用有机溶剂甲醇配置的,这两个样进行液相色谱分析后的峰有区别吗?我的流动相是甲醇-乙腈-草酸谢谢!







 我要推广仪器
我要推广仪器
 下载APP
下载APP