柑橘和土壤中四螨嗪残留量的液相色谱测定法摘要: 目的 用液相色谱测定柑橘和土壤中四螨嗪的农药残留量。方法 样品用丙酮提取,干装硅胶柱净化,利用安捷伦 HPLC 1200液相色谱进行检测。结果 四螨嗪农药的检出限为0. 01 mg /kg ,土壤的添加回收浓度在0. 05 ~ 2. 03mg /kg时,平均回收率为93. 1% ~ 95. 8%,相对标准偏差为0. 30% ~ 5. 34%,柑橘的添加回收浓度在0. 05 ~ 2. 01 mg /kg 时,平均回收率为92% ~ 95. 4%,相对标准偏差为0. 33% ~ 4. 60%。
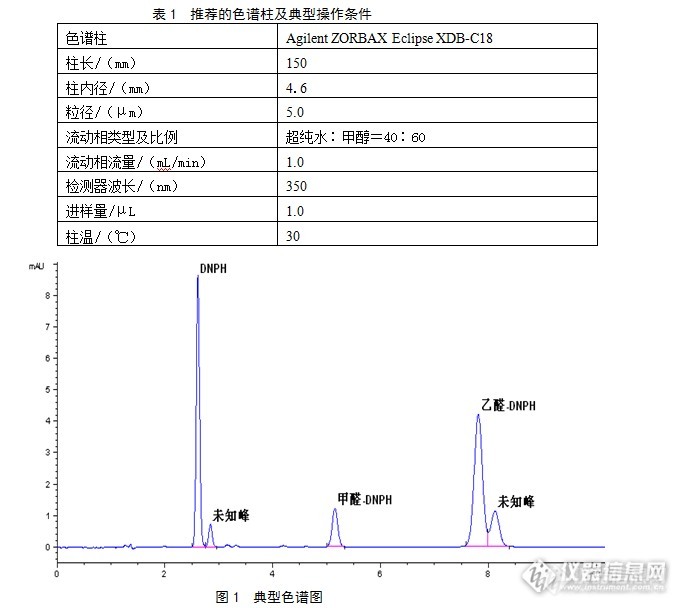
微量甲醛、乙醛液相色谱测定法1 适用范围本标准规定了液相色谱测定水溶液或环氧乙烷样品中的甲醛和乙醛的方法。本标准适用于样品中甲醛、乙醛含量为1~50mg/kg的分析。2 方法概要 液态样品中微量甲醛、乙醛与2、4-二硝基苯肼衍生生成甲醛-2、4-二硝基苯腙(以下称:甲醛-DNPH)和乙醛-2、4-二硝基苯腙(以下称:乙醛-DNPH),在通过固相C18小柱将甲醛、乙醛的衍生物从溶液中萃取出来。随后,使用自动进样器直接将萃取出的甲醛、乙醛衍生物注入[url=https://insevent.instrument.com.cn/t/Mp]气相色谱仪[/url],样品在甲醇和水混合溶液携带下通过毛细管色谱柱分离,使样品里的甲醛衍生物、乙醛衍生物及未反应2、4-二硝基苯肼按照不同顺序先后进入紫外检测器检测。最后,由色谱数据工作站采用外标法得到各物质含量。3 试剂和材料3.1 甲醇:液相色谱(HPLC)纯。3.2超纯水:比电阻率大于18.2ΜΩcm的水。3.32,4-二硝基苯肼(DNPH):分析纯。3.4超声波振荡器。3.5玻璃溶剂过滤器。3.6真空泵。3.7量筒:10mL。3.8量筒:25mL。3.9具塞碘量瓶:250mL。3.10C18固相萃取小柱(SPE)。3.11注射器:5mL。3.12注射器:10mL。3.13烧杯:50mL3.14标准溶液:与分析样品浓度相接近甲醛、乙醛水溶液,自配或外购。4 仪器4.1液相色谱仪:安捷伦科技公司1200型,配有自动进样器、真空脱气、四元梯度泵和紫外检测器,或同类产品。4.2色谱数据工作站:安捷伦科技公司Chemstation工作站,或同类产品。4.3色谱柱:见表1推荐色谱柱,或同类产品。推荐色谱柱典型操作条件见表1,典型色谱图见图1。[img=,681,608]http://ng1.17img.cn/bbsfiles/images/2017/09/201709032112_01_2166779_3.png[/img]5 校正仪器第一次使用或者被改变了某一或某些参数设置(如流量、流动想类型、流动相比例、色谱柱等),应对仪器进行校正。使用已知与样品接近含量的标准甲醛、乙醛溶液与DNPH络合后,在液相色谱仪上分析,测量两物质色谱峰峰面积,计算校正因子k[sub]i[/sub]:k[sub]i[/sub]=标准液中i物质浓度/i物质衍生物峰面积。6 试验步骤6.1用5mL注射器将5mL甲醇注入C18固相萃取小柱,冲洗萃取小柱进行预处理,然后再用5mL超纯水冲洗。6.2分别取10g样品(由于环氧乙烷沸点较低,环氧乙烷产品可直接使用量筒量取11.2mL)、10mL2,4-二硝基苯肼(DNPH)试剂和10mL超纯水转移到碘量瓶内,盖上瓶塞,让混合物反应15分钟。如果观察到沉淀,则应使用超纯水按1:10稀释样品,并按上述描述再次进行操作。6.3使用一个10mL量筒量取10mL的反应混合物,用针筒将其转移到一个6.1中预处理好的固相萃取小柱,注射时应将样品缓慢地推进萃取小柱。然后,使用10mL超纯水重复上述步骤,注意需将所有液体推出萃取小柱。6.4用注射器将8mL甲醇推进萃取小柱,以洗脱富集被固相萃取小柱从样品中萃取出的甲醛-DNPH、乙醛-DNPH及未反应的DNPH溶液。洗脱富集液放置于烧杯中。再向烧杯添加少量甲醇,使溶液的最终重量达到10±0.01g。6.5使用符合4.3中推荐色谱柱或同类色谱柱,采用自动进样器将适量6.4中的洗脱富集样品注入4.1规定液相色谱仪,通过色谱工作站测量各物质出峰面积,利用已测校正因子面积归一定量计算样品中各组成含量。6.6用10mL的去离子水替代样品重复步骤6.2至6.5,测定空白。7 计算7.1溶液甲醛、乙醛含量按下式计算:(外标法)X[sub]i[/sub]=A[sub]i[/sub]×k[sub]i[/sub]式中:X[sub]i[/sub]——被测组分i的含量,mg/kg;A[sub]i[/sub]——被测组分i的衍生物峰面积;k[sub]i[/sub]——被测组分i的绝对校正因子;i——甲醛或乙醛。8 精密度8.1 重复性同一操作者重复测定的两次测定结果的差值应不超过平均值的10%(m/m)。9 结果的表示仪器稳定状态下连续两次测定结果的平均值作为出厂产品的测定结果,生产装置过程控制使用单次测定数据作为结果报告,结果的表示修约至1mg/kg。10注意事项10.1 仪器进样前取样,需摇晃采样容器,以保证分析样品的均匀性。10.2 色谱流动相必须使用液相色谱(HPLC)纯试剂。10.3当分析环氧乙烷或含有环氧乙烷样品时,使用前应事先将试剂、容量瓶和量筒冷却,以防止环氧乙烷受热挥发。10.4 对含有环氧乙烷的样品的任何操作,必须在通风柜中完成,并做好个人防护。
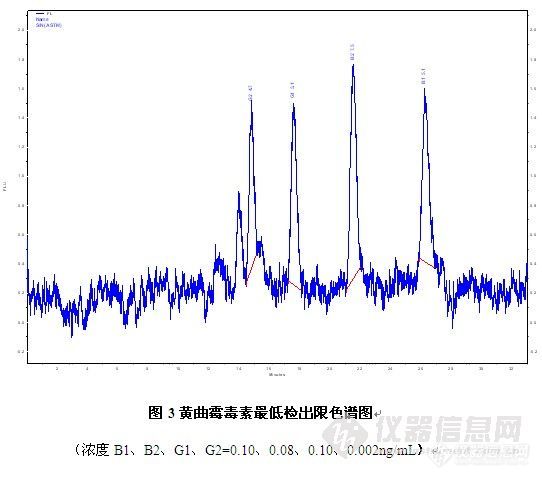
维权声明:本文为xiaoyuer原创作品,本作者与仪器信息网是该作品合法使用者,该作品暂不对外授权转载。其他任何网站、组织、单位或个人等将该作品在本站以外的任何媒体任何形式出现均属侵权违法行为,我们将追究法律责任本文提供黄曲霉毒素柱后光衍生荧光测定法,该方法相对柱后试剂衍生操作更简单,同时提高黄曲霉毒素B1、G1的灵敏度。1仪器与试剂HITACHI L-2000高效液相色谱仪、配L-2485荧光检测器,柱后光化学衍生装置,各种试剂均为色谱纯或分析纯,实验用水为超纯水,免疫亲和柱Alfa Test(VICAM),黄曲霉毒素对照品均为SIGMA标准品。样品为市售狗粮。2色谱条件色谱柱:Hitachi LaChrom C18 (5μm)4.6mmI.D.x250mmL.流动相:CH3OH:H2O =45:55流速:0.8mL/min柱温:30℃波长:Ex:365nm Em:435nm进样体积:20µL3样品前处理方法同反相液相色谱荧光直接测定法,样品过免疫亲和柱后,过滤膜测定。4 结果与讨论4.1标样、样品的测定在选定的条件下四种常测黄曲霉毒素得到很好的分离,通过光柱后衍生后,黄曲霉毒素B1、G1灵敏度比直接测定大幅提高。http://ng1.17img.cn/bbsfiles/images/2010/11/201011181538_260460_2961690_3.jpg4.2灵敏度比较 相同浓度的黄曲霉毒素混合标样,仪器接柱后光化学衍生装置,图2中A为衍生装置未打开得到的色谱图,B为装置开启得到的色谱图,比较A[s
求助!色谱条件与测定法照Y-SOP-QC-T-091《高效[url=https://insevent.instrument.com.cn/t/5p][color=#3333ff]液相色谱[/color][/url]法标准操作规程》测定。色谱柱:以十八烷基硅烷键合硅胶为填充剂(推荐使用Agilent Eclipse XDB-C184.6mm×150mm5μm)。检测波长:254nm 流速:l.0 mL/min(必要时可调节);柱温:30℃:样品盘温度:7℃;进样量:20ul 流动相:以三乙胺醋酸溶液(取三乙胺14ml与冰醋酸5.7ml,置100ml量瓶中,加水稀释至刻度,摇匀)-乙腈-水(1.2:120:880)并用冰醋酸调节pH值至3.0±0.2。总量配15000ml 三乙胺应该加多少??急急急
高效液相色谱法系统适用性试验拖尾因子(T)规定峰面积测定法时,T值偏离过大,也会影响检测和定量的准确性。请问各位T值有没有一个范围?[em0805] [em0805]
【含量测定】 照高效液相色谱法(附录24页)测定。色谱条件与系统适用性试验 用十八烷基硅烷键合硅胶为填充剂,0.025 mol/L磷酸溶液-乙腈(87:13)用三乙胺调节pH值至3.0为流动相,检测波长为277nm。理论板数按环丙沙星峰计算应不低于2000,环丙沙星峰与内标物质峰的分离度应符合要求,拖尾因子应不大于2.5。内标溶液的制备 取恩诺沙星对照品25 mg,精密称定,置100ml量瓶中,加流动相溶解并稀释至刻度,摇匀,即得。测定法 取乳酸环丙沙星对照品约25 mg,精密称定,置100ml量瓶中,加流动相溶解并稀释至刻度,摇匀;精密量取该溶液与内标溶液各5ml,置25ml量瓶中,用流动相稀释至刻度,摇匀,取20µ l注入液相色谱仪,记录色谱图;另取本品适量,同法测定。按内标法以峰面积计算,结果与0.7863相乘,即得。
维权声明:本文为xiaoyuer原创作品,本作者与仪器信息网是该作品合法使用者,该作品暂不对外授权转载。其他任何网站、组织、单位或个人等将该作品在本站以外的任何媒体任何形式出现均属侵权违法行为,我们将追究法律责任。黄曲霉毒素的测定国家标准方法有很多种,有采用柱后试剂衍生反相液相色谱荧光法、柱前衍生反相液相色谱荧光法、酶联免疫吸附法、薄层法,柱后试剂衍生需要添加柱后衍生用液相泵、反应盘管等,柱前衍生重现性相对较差且操作麻烦,酶联免疫及薄层相对准确性较差,本文采用常规四元梯度反相液相色谱配L-2485荧光检测器直接测定黄曲霉毒素,操作简单、灵敏度较高。1仪器与试剂HITACHI L-2000高效液相色谱仪、配L-2485荧光检测器,各种试剂均为色谱纯或分析纯,实验用水为超纯水,免疫亲和柱Alfa Test(VICAM),黄曲霉毒素对照品均为SIGMA标准品。样品为市售玉米渣。2色谱条件色谱柱:Hitachi LaChrom C18 (5μm)4.6mmI.D.x250mmL.流动相:CH3OH:H2O =45:55流速:0.8mL/min柱温:30℃波长:Ex:365nm Em:435nm进样体积:20µL3样品前处理准确称取25克样品加入125mL甲醇水(7:3)溶液,加入5克氯化钠,振荡溶液30min,纤维滤纸过滤,移取10mL滤液,加入10mL水,将溶液过免疫亲和柱,用20mL超纯水淋洗,用真空泵抽干柱,加1mL甲醇溶液洗脱,收集洗脱液,加水1mL,过0.45μm滤膜,作为待测样。4 结果与讨论[font='Tim

高效液相色谱法测定纺织品中甲醛含量的方法验证摘要:采用2,4-二硝基苯肼为衍生化试剂,生成2,4-二硝基苯腙,用高效液相色谱-二极管阵列检测器(HPLC-DAD)测定纺织品萃取后有颜色干扰的样品效果良好。方法检出限小于5.0mg/kg;从0.15 mg/L起到7.50mg/L的范围(样品浓度对应为7.5mg/kg到375mg/kg的范围)内成线性且相关系数均在0.999以上;组内精密度及组间精密度分别为1.51%及3.50%,表面方法稳定性良好;采用不同浓度的加标回收率实验,回收率落在85%-110%,RSD在5%以内,方法测试结果良好。本方法可以很好的用于纺织品甲醛含量的检测,尤其是对有颜色干扰的样品萃取液有检测优势。关键词:纺织品;甲醛;高效液相检测法;颜色干扰。1.前言 目前,甲醛的检测方法按精确度划分,大致可分为两种,其一种为精密度测定法(仪器分析法),包括世界卫生组织推荐的高效液体色谱法(HPLC),气相色谱法(DNPH-GC法)及分光光度法等;其二为简易测定法,该法主要用于快速检测,其精确度要求不高。主要有电法学方法,可以显示测定数据,以及检测管方式和测定纸方式,即通过检测气体与指示剂发生法学反应而表现出的颜色变化来测定检测气体浓度。 目前纺织品甲醛的检测方法主要有GBT 2912.1-2009 纺织品甲醛的测定第1部分游离和水解的甲醛(水萃取法)(MOD 14184-1-2008);GBT 2912.2-2009 纺织品甲醛的测定第2部分:释放的甲醛(蒸汽吸收法) (MOD 14184-2-2008);ISO14184-1-2011textiles-determination of formaldehyde part1:free and hydrolysed formaldehyde(waterextraction method);ISO14184-2-2011textiles-determination of formaldehyde part2:released formaldehyde(vapourabsorption method);JIS L1041-2011-树脂整理织物试验方法;GB/T 2912.3-2009 纺织品甲醛的测定第3部分:高效液相色谱法。大部分方法都还是采用乙酰丙酮显色法,[font='Calibri','sans-s
请问有人知道液相色谱法中测甘氨酸的拖尾因子偏低是什么原因导致的? 我这里是用药典通则3123 人免疫球蛋白中甘氨酸含量测定法来测定甘氨酸含量。[url]http://db.ouryao.com/yd2015/view.php?v=txt&id=5543[/url],药典的要求,系统适用性拖尾因子要在0.95-1.40范围内。 但是我这里做出来的拖尾因子为0.95,有时候会是0.94。流动相的PH为5.08,想问下各位有没有人知道是什么原因导致的。 因为我们是用峰面积来计算的,所以其实拖尾因子偏低一点,但是只要不是偏离的太厉害,我觉得其实是没问题的,所以,如果拖尾因子实在是达不到,请问可以将药典规定的0.95-1.40这个范围降低吗?

【作者】:赵娜萍, 余露山, 姚彤炜, 曾苏【摘要】:目的研究帕洛诺司琼的体外代谢,建立人肝微粒体中帕洛诺司琼的反相高效液相色谱测定法。方法帕洛诺司琼与人肝微粒体共孵育之后用乙醚提取,采用地非三唑为内标,以DiamonsilC18柱(4.6mm×200mm,5μm)为分析柱,0.01μmol.L-1KH2PO4(pH3.0)-甲醇(30∶70)为流动相,流速1.0mL.min-1,紫外检测波长为241nm。结果帕洛诺司琼在1~100μmol.L-1内线性关系良好(r=0.9999,n=5),检测限为0.05μmol.L-1(S/N≥3),定量限为(0.21±0.04)μmol.L-1(n=5)。方法提取回收率和方法回收率平均分别为89.7%和100.0%,日内,日间RSD均10%(n=5)。结论此法简便,准确,可用于研究帕洛诺司琼在人肝微粒体中的代谢。 【作者单位】:浙江大学药学院药物分析与药物代谢研究室; 浙江大学药学院药物分析与药物代谢研究室 杭州310058; 杭州310058;【关键词】:帕洛诺司琼; 高效液相色谱法;http://ng1.17img.cn/bbsfiles/images/2012/09/201209022110_388001_1838299_3.jpg
药典方法示例:[b]黄曲霉毒素测定法[/b]混合对照品溶液的制备:精密量取黄曲霉毒素混合标准品(黄曲霉毒素B1、黄曲霉毒素B2、黄曲霉毒素G1、黄曲霉毒素G2标示浓度分别为1.0μg/ml、0.3μg/ml、1.0μg/ml、0.3μg/m1)0.5ml,置10ml量瓶中,用甲醇稀释至刻度,作为储备液。精密量取储备液1ml,置25ml量瓶中,用甲醇稀释至刻度,即得。分别精密吸取上述混合对照品溶液5μl、10μl、15μl、20μl、25μl,注入液相色谱仪,测定峰面积,以峰面积为纵坐标,进样量为横坐标,绘制标准曲线。[b]硫酸依替米星[/b]第二法 照高效液相色谱法(通则 0512)测定测定法:取依替米星对照品适量,精密称定,分别加水溶解并定量稀释制成每1ml中约含依替米星1.0mg、0.5mg和0.25mg的溶液作为对照品溶液(1)、(2)、(3)。精密量取上述三种溶液各20μl,分别注入液相色谱仪,记录色谱图,以对照品溶液浓度的对数值对相应的峰面积的对数值计算线性回归方程,相关系数(r)应不小于0.99;[b]蜂蜜[/b]标准曲线的制备:分别精密称取果糖对照品1.0g,葡萄糖对照品0.8g,置同一具塞锥形瓶中,精密加入40%乙腈20ml,溶解,摇匀,作为果糖、葡萄糖对照品储备液。另精密称取蔗糖对照品0.2g,麦芽糖对照品0.2g,置同一具塞锥形瓶中,精密加入40%乙腈10ml,溶解,摇匀,作为蔗糖、麦芽糖对照品储备液。分别精密量取果糖、葡萄糖对照品储备液和蔗糖、麦芽糖对照品储备液,加40%乙腈配成不同浓度的果糖、葡萄糖、蔗糖、麦芽糖混合对照品溶液。精密吸取混合对照品溶液各15μl,注入液相色谱仪,分别测定。以对照品浓度为横坐标,以峰面积值为纵坐标,绘制标准曲线,计算回归方程。[b]制作方法总结如下:[/b]1.称一份对照品制备作为对照品储备液;2.稀释成系列梯度浓度,取相同的进样量上机检测或配制一份对照品溶液取不同的进样量。 有一个系统风险在里面,就是对照品如果称量不准确,整个标准曲线就不准确。一般外标法都要求配制两份标准溶液,标准曲线法也应该配制两份标准溶液,一份用于制作标准曲线,另一份标准溶液用于标准曲线的校验,以减少系统风险。最准确的方法应该是每一个标准浓度点都应从称量开始,但也是成本最高的方法。
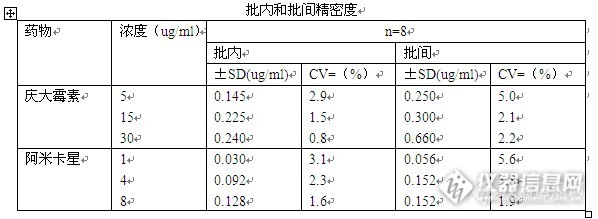
高效液相色谱法快速测定血清中的庆大霉素和阿米卡星庆大霉素和阿米卡星是用于严重革兰氏阴性细菌感染治疗的氨基糖苷类抗生素。像其他氨基糖苷类抗生素一样,庆大霉素和阿米卡星的有效治疗范围狭窄容易导致肾脏毒性和耳毒性。因此,监测庆大霉素和阿米卡星血清中的水平对于安全和有效的临床应用十分重要。最近,高压液相色谱法已被报道用于氨基糖苷类抗生素的测定。多数测定需要柱前或柱后衍生荧光检测。这些方法涉及耗时的预处理,如血清中氨基糖苷类抗生素的柱萃取以及溶剂。本实验开发了一种较为简单的预处理方法来测定血清中的庆大霉素和阿米卡星,并与荧光偏振免疫测定法进行了比较。材料和方法:庆大霉素和阿米卡星购自药店,邻苯二甲醛(瑞尔丰化工),2-巯基乙醇、庚烷磺酸钠、1,2-乙烷二磺酸二钠、色谱乙腈、蒸馏水。蛋白质沉淀剂(10mM辛烷磺酸钠溶解于3.5%高氯酸中)。庆大霉素的流动相为含有22 mM的1,2 - 乙烷二磺酸二钠和5mM辛烷磺酸钠的水 - 乙腈混合物(80:20,体积/体积),用乙酸调节pH至约3.5。阿米卡星的流动相为含有37 mM的1,2 - 乙烷二磺酸二钠和5mM辛烷磺酸钠的水 - 乙腈混合物(80:20,体积/体积),用乙酸调节pH至约3.5。仪器和色谱条件:安捷伦高效液相色谱仪1200,UV检测器,色谱柱:安捷伦Agilent液相色谱柱4.6﹡150﹡5u,流动相的流速保持在1毫升/分钟。邻苯二甲醛试剂用泵以0.6毫升/分钟的流速。实验部分:将50ul血浆加入到50ul蛋白质沉淀剂,涡旋混合数秒后,使用KM-15200离心机离心2分钟(15000rpm),离心后上清液进样。正常血清,分别加入已知量的庆大霉素和阿米卡星,进行分析,根据加入量和峰面积绘制标准曲线。荧光偏振免疫通过市售的试剂盒进行。结果:图1为庆大霉素的标准色谱图,庆大霉素加入对照血清的色谱图以及对照血清的色谱图。(其中交标庆大霉素的含量为15ug/ml)http://ng1.17img.cn/bbsfiles/images/2014/09/201409300949_516466_2188679_3.jpg图2为阿米卡星的标准色谱图,阿米卡星加入对照血清的色谱图以及对照血清的色谱图,经过蛋白质沉淀剂处理后的血清和未处理的色谱图基本形同,皆未见到影响抗生素检测的干扰杂质峰。http://ng1.17img.cn/bbsfiles/images/2014/09/201409300949_516467_2188679_3.jpg以峰面积作为纵坐标,血清中的药物浓度为横坐标得到庆大霉素的线性标准曲线(3-50ug/ml)为y =0.979x - 0.006,阿米卡星的线性标准曲线(0.5-10ug/ml)为y= 0.998x - 0.04根据庆大霉素和阿米卡星在血清中不同浓度的重复测定,得到庆大霉素和阿米卡星的批内变异系数分别为0.8 -2.9%和1.6-3.1%,批间变异系数为2.0-5.0%及1.9-5.6%。http://ng1.17img.cn/bbsfiles/images/2014/09/201409300949_516468_2188679_3.jpg回收率试验:分别将15ug/ml的庆大霉素水溶液加入含有15ug/ml阿米卡星的血清中和将4ug/ml的阿米卡星水溶液加入4ug/ml的血清中。将它们的峰面积进行比较。基于三个样品的平均值,庆大霉素和阿米卡星的分别为99.0%和98.5%.通过该方法获得的结果与用荧光偏振免疫测定法进行了比较,回归方程和相关系数分别为庆大霉素:y=1.027x - 1.090,n =44和r=0.996。阿米卡星y=0.998x - 0.206, n =42和r=0.957。讨论:本实验采用了蛋白质沉淀剂,排除干扰物,再将样品进行分析,优化的分析方法与荧光偏振免疫测定法进行了比较种,该方法方法的优点是速度快,操作简便,重复性好。因此,该方法可用于该药物的分析。
针对近期奶中出现的黄曲霉毒素M1超标问题提出以下液相色谱法精准检测方案。1. 仪器1.1 高效液相色谱仪配荧光检测器1.2震荡器(或高速搅拌器)1.3 高速离心机1.4 泵流操作架(带气泵)2. 试剂与试液2.1 色谱甲醇2.2 蒸馏水2.3 乙腈+水(25+75)2.4 石油醚2.5 10%乙腈2.6 氢氧化钠溶液(0.5mol/L)玻璃纤维滤纸(PriboFast免疫亲和柱免费赠送)3. 测定法本法系用高效液相色谱法(GB/T 18980-2003)测定牛奶,奶粉等产品中的黄曲霉毒素M1,除另有规定外,按下列方法测定。3.1 色谱条件色谱柱:C18柱,150×4.6mm,5um检测器:荧光检测器;激发波长365nm,发射波长435nm;流动相:乙腈-水(25:75)流速:1.0ml/min进样量:20ul柱温:室温3.2 标准品溶液的配制取黄曲霉毒素M1标准品,用乙腈稀释成0.5ug/ml的标准储备液。根据使用需要,准确吸取10μl、20μl、50μl、1
高效液相色谱定量分析1 黄芩苷含量检测(1)色谱条件与系统适应性试验 以十八烷基硅烷键合硅胶为填充剂;流动相:甲醇-水-磷酸(47:53:0.2);检测波长:276nm。柱温:30℃;流速:1mL/min。理论板数按黄芩苷峰计算应不低于4500。(2)测定方法对照品溶液的制备 取黄芩苷对照品适量,精密称定,置棕色量瓶中,加50%甲醇适量,置水浴中振摇使溶解,放置至室温,稀释至刻度,摇匀,制成每1mL含40μg的溶液,即得。 供试品溶液的制备 精密量取样品溶液,置容量瓶中,加水稀释至一定浓度,摇匀,用0.45μm滤头过滤即得。测定法 分别精密吸取对照品溶液10μL与供试品溶液10μL,注入色谱仪,测定,即得。2 高效液相色谱的定量分析药典规定:本品每支10mL含黄芩苷(C21H18O11)计,不少于80mg。本品每1mL含金银花以绿原酸(C16H18O9)计,不得少于0.60mg。(1)黄芩苷高效液相图 见附件1 [img]http://www.instrument.com.cn/bbs/images/affix.gif[/img][url=http://www.instrument.com.cn/bbs/download.asp?ID=101403]附件1 [/url]
双癸基二甲基氯化铵是一种阳离子表面活性剂。分子式是C22H48ClN,相对分子质量是362.08,属双链季铵盐类化合物,常温下为淡黄色透明液体,易溶于水和有机溶剂,前些天,客户拿了一个文献《复方消毒液中双癸基二甲基氯化铵的高效液相色谱测定法》,方法是采用C18色谱柱子,ELSD检测器,今天测试了后发现效果不行。几个问题:1、你觉得这个物质用ELSD这样测可行吗?2、我应该如何来控制保留时间呢?3、这个组分是不是对色谱柱侵蚀超强啊?
建立了牛奶中4 种安乃近代谢物—— 4- 甲酰氨基安替比林、4- 乙酰氨基安替比林、4- 氨基安替比林和4- 甲基氨基安替比林的液相色谱- 串连质谱(LC-M/MS)测定法。样品加入TRIS 缓冲溶液后用乙腈提取,提取液经乙腈饱和过的正己烷脱脂净化,采用[url=https://insevent.instrument.com.cn/t/Yp][color=#3333ff]LC-MS[/color][/url]/MS 电喷雾正离子(ESI+)、多反应监测(MRM)模式检测,4- 乙酰氨基安替比林、4- 甲酰氨基安替比林和4- 氨基安替比林采用外标法定量,4- 甲基氨基安替比林采用内标法进行定量。4- 甲酰氨基安替比林的检出限为0.24μg/kg,4- 氨基安替比林的检出限为0.59μg/kg,4- 乙酰氨基安替比林的检出限为0.20μg/kg,4- 甲基氨基安替比林的检出限则为0.61μg/kg。在添加量5~20μg/kg 范围内,4 种安乃近的回收率在80.4%~97.9% 范围内,相对标准偏差(RSD)均小于9%。
[color=#444444]最近想建一个物质的检测方法,找了其类似物-苯乙酸甲酯的含量测定的相关方法,文献上说“按酯测定法(OT-18)中的方法一测定。所取试样量为1 g。计算中的当量因子(e)取75.09。或按[url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]法(GT-10-4)中用非极性柱方法测定。” 但怎么样也找不到酯测定法(OT-18)和[url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]法(GT-10-4)的相关资料,请问哪位能帮我找一下这方面的资料吗? 谢谢!!![/color]
10-羟基-α-癸烯酸是脂肪酸的一种,蜂王浆所含的重要物质,也称王浆酸,简称10-HDA,是至今发现仅在王浆中天然存在的有机烯酸。10-羟基-α-癸烯酸也是目前测定蜂王浆的含量指标,保健食品中常用的检测方法是高效液相色谱测定法。而标准方法也只有一个《SN/T 0854-2000 进出口蜂王浆及蜂王浆冻干粉中10-羟基-α-癸烯酸检验方法》,液相方法用内标的并不多,这个方法就是其中之一,我一直也不明白为什么要内标,因为样品的处理方法并不是很复杂,有必要加内标吗?由于没有内标,所以我在测试的时候也就没有加内标。以前是同事做的多,我做这个也就一两次吧,虽然没什么经验,都是我还是以分享为主,让大家都能参与到讨论中来,下面就简单分享下我做的过程吧。色谱柱:Topsil 液相色谱柱(C18,5um,4.6*250mm)检测波长:210nm流动相:甲醇+0.01mol/L HCL=55+45流速:1.0mL/min样品处理:1. 粉末状固体样品:称取样品后,加2 mL 0.01mol/L HCL,摇匀后加甲醇,超声溶解过滤2. 液体样品:用稀盐酸调PH到2-3,用甲醇定容超声溶解3. 油状样品:加几滴30%氢氧化钠和少量水溶解,用二氯甲烷提取后分取二氯甲烷低温蒸干,残渣用甲醇溶解这个图是我做的标准色谱图http://ng1.17img.cn/bbsfiles/images/2011/12/201112211401_340007_1608710_3.gif这个是样品色谱图http://ng1.17img.cn/bbsfiles/images/2011/12/201112211402_340008_1608710_3.gif总结:1. 如果是样品类型多样,可以根据样品来选择合适处理方法,样品处理加盐酸与不加结果相差不大,加盐酸的结果高一些。2. 保健食品中蜂王浆冻干粉的配方剂型较多,一般情况下可以直接用甲醇超声提取3. 根据样品的配方组成及色谱柱的情况,可适当调整流动相的比例。4. 其实流动相还可以用磷酸水溶液,峰形也很不错,我用这个流动相,只是参照了进出口标准方法的流动相。你有什么色谱实验可以分享吗?别等啦,原创大赛马上就结束咯哟~~~
按照2010新版药典法进行的测定,是新药典中典型的由有关物质测定由等度变梯度的实例,也是典型的含量测定方法由05版的电化学滴定法变液相色谱法实例。
请问如何用液相色谱测定阴离子。我液相色谱是VWD,应用何种色谱柱,用何种流动相?
采用2010版药典方法测定,有关物质检测由等度变梯度,含量测定由电位滴定法变液相色谱法。
能不能用液相色谱法?我做的是大鼠的大脑海马细胞,想用液相色谱法测定一下细胞培养液上清中的乙酰胆碱,GABA ,谷氨酸。请问能用液相色谱法测吗?
照Y-SOP-QC-T-091《高效[url=https://insevent.instrument.com.cn/t/5p][color=#3333ff]液相色谱[/color][/url]法标准操作规程》测定。色谱柱:以十八烷基硅烷键合硅胶为填充剂(推荐使用Agilent Eclipse XDB-C184.6mm×150mm5μm)。检测波长:254nm 流速:l.0 mL/min(必要时可调节);柱温:30℃:样品盘温度:7℃;进样量:20ul 流动相:以三乙胺醋酸溶液(取三乙胺14ml与冰醋酸5.7ml,置100ml量瓶中,加水稀释至刻度,摇匀)-乙腈-水(1.2:120:880)并用冰醋酸调节pH值至3.0±0.2。总量配15000ml,三乙胺应该加多少?
[b][color=Blue]一般来讲,不管是哪种测定方法,测定的结果应该基本一致但在采用[url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]及液相色谱分析同一样品时发现,[url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]及液相色谱均已进行了方法学考察,且均获得很好的结果(包括精密度、准确度、重复性等等)后,两种方法测定结果竟然能有10%的差异,是和道理?[/color][/b]
不溶性糖精的高效液相色谱测定方法?请问用高效液相色谱法测定不溶性糖精的具体测量条件,如流动相、流速等,越详细越好,谢谢!!!!
农药悬浮剂的提取5%联苯菊酯悬浮剂,选用液相色谱测定,物质峰有干扰,采用C18固相萃取柱净化,效果良好但回收率低,有没有其他净化方法?
[img]http://www.instrument.com.cn/bbs/images/affix.gif[/img][url=http://www.instrument.com.cn/bbs/download.asp?ID=30656]SH/T 0714-2002 石脑油中单体烃组成测定法(毛细管[url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]法)[/url]
[size=3][b]儿茶酚胺测定[/b][/size]最近采用液相色谱-电化学检测器测定血浆和尿中的儿茶酚胺,不知各位有否这方面的经验,能否相互交流一下样品预处理条件和色谱条件,谢谢!
用液相色谱测定水中的菲,需要什么前处理,样品中有机物杂质含量低,只需定量.用什么检测器,我是菜鸟,请高手指点!谢谢![em0808][em0808]
阳离子树脂 预处理 测氨基酸用液相色谱测定湖水中氨基酸,先过阳离子树脂交换柱,去除钙、镁离子的影响,请问预处理步骤是什么?是否需要索氏浸提树脂?谢谢!







 我要推广仪器
我要推广仪器
 下载APP
下载APP