
以前一位版友在某XRD的QQ群上问怎样在全谱拟合软件中扣除Kα2我很不解。。。既然都是全谱拟合了。。。Kα2都已经拟合进去了。。。为什么还要修改原始数据去扣除Kα2呢?昨天我终于得到一个让人信服的理由:某些做PDF分析的同志需要对单色的衍射数据进行傅里叶转化然后用PDF分析软件处理数据。。。于是就有了这个“用TOPAS全谱拟合法扣除Kα2”:1. 对原始谱进行全谱拟合,包含Kα1,Kα2,K其他。。。:http://ng1.17img.cn/bbsfiles/images/2015/05/201505060302_544905_1986542_3.jpg2. 固定所有精修变量,将Kα1的面积改为0,再计算一次谱(实际上是用除了Kα1以外的所有波长计算谱)http://ng1.17img.cn/bbsfiles/images/2015/05/201505060303_544906_1986542_3.jpg3. 导出残差数据和背景线到Excel,两者相加就是只包含Kα1的衍射谱了。。。http://ng1.17img.cn/bbsfiles/images/2015/05/201505060303_544907_1986542_3.jpg是不是很简单丫?http://simg.instrument.com.cn/bbs/images/default/em09503.gif
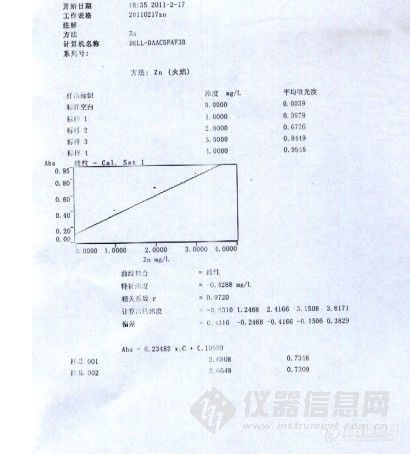
[size=3][b]瓦里安[url=https://insevent.instrument.com.cn/t/Wp][color=#3333ff]原吸[/color][/url]“线性”和“新合理”之间的区别。[/b][/size]新手提问,我想问一下瓦里安AA240中曲线拟合法中“线性”和“新合理”有什么区别。同几瓶标液用“线性”做出相关系数R只有0.9720,但是“新合理”做出来的相关系数R却是1.000,这之间也太蹊跷了吧。(当时瓦里安装机人员是要我最好用“线性”,但是之后有人说“新合理”做也是一样的。) 新合理是什么具体我也说不上,应该说是曲线拟合法中的一种方法。不是两个点,也是三到四个点。“线性”曲线图显示的几个点在一条直线上,新合理的曲线图则可能是一条抛物线。 可以看附件图片。第一张为线性R0.9720,第二张而相同的几个标液在新合理中相关系数R1.000,曲线成抛物线状态。第三张图片为新合理相关系数R1.000,但是曲线为直线。[img]http://ng1.17img.cn/bbsfiles/images/2017/01/201701191653_630401_2163539_3.jpg[/img][img]http://ng1.17img.cn/bbsfiles/images/2011/02/201102191711_278481_2163539_3.jpg[/img][img]http://ng1.17img.cn/bbsfiles/images/2011/02/201102191711_278482_2163539_3.jpg[/img]
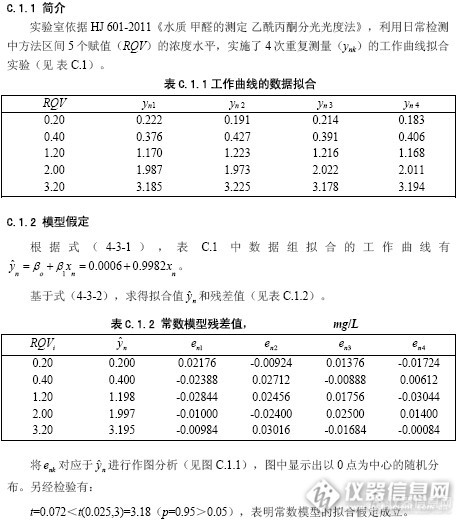
CNAS-GL34,线性拟合法评定不确定度在第28页有相关举例,但第28页最后一行的t值,未给出计算公式,有哪位知道这是如何计算的吗,是用什么公式算的?
各位老师好,最近在使用内标法分析转化率,但是做出的标准曲线用直线拟合比较差,请问这个可以用吗,出现这种情况是什么原因呢?待测物质足够稀释,应该不存在溶液饱和,色谱的峰也是标准对称峰。
最近做了一些荧光粉样品。可是不会用Jade 5软件通过全谱拟合法得到新的晶胞参数。主要有以下几个问题:一,检测的物像不只一个,这时还能得到晶胞参数吗?二,用手动拟合时,R值在20%以上,这样得到的晶胞参数可靠嘛?三,如果直接点全谱拟合,再占cell refine,得到的晶胞参数是不是就是该样品的晶胞参数,
就是将几个合在一起的峰拟合成独立的峰,还是需要单独购买这样的软件?
今天实验室一MM问我,瓦里安的曲线拟合方式中的新合理的二次曲线是怎么个拟合法的,我也整不明白,有专家解释下吗
我用高效液相色谱测银杏内酯A B,如果根据峰面积和浓度线性拟合的话,峰面积重复性不好。想问大侠一下,高效液性色谱可以用峰高和浓度线性拟合,得到线性拟合直线吗?求大侠支招,不胜感激
[color=#444444][url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]外表定量时,做峰面积与浓度标线时,可以用二次拟合吗?为什么标准曲线都是线性的,不用多项式拟合?[/color]
[color=#444444]我用高效液相色谱测银杏内酯A B,如果根据峰面积和浓度线性拟合的话,峰面积重复性不好。想问大侠一下,高效液性色谱可以用峰高和浓度线性拟合,得到线性拟合直线吗?求大侠支招,不胜感激[/color]
[color=#444444]我用高效液相色谱测银杏内酯A B,如果根据峰面积和浓度线性拟合的话,峰面积重复性不好。想问大侠一下,高效液性色谱可以用峰高和浓度线性拟合,得到线性拟合直线吗?求大侠支招,不胜感激[/color]
用NMR得到了C4区的谱图。现在要对这条谱线进行光谱拟合,就是要用高斯函数和洛仑兹函数的混合函数对它进行拟合。有模型但我不知道该如何导入这个混合函数,用过origin和peakfit等软件,请问这种情况该如何处理?谱图及欲得到的拟合结果见附件
我用opus拟合红外图谱,需要求拟合出来的峰值的面积,怎么求呢?是那个报告中的 “积分”的值吗?
用jade里的手动拟合时,是只对和卡片匹配的峰进行拟合,还是对所有峰进行拟合?拟合时很小的峰(分辨不出真假)也要拟合吗?拟合的结果全铺上面那个曲线越平滑越好吗?另外一个问题:用jade自身带的晶粒尺寸计算工具计算的结果和谢乐公式计算的结果还有电镜观察的结果是不是应该大体一致?
在做图谱拟合的时候要求R小于9%,但是我无论怎么拟合,R都在10以上.请问是不是因为我的XRD衍射图谱不是纯物质引起的呢?请有经验的朋友指教!
请问使用Mestrenova软件对固体核磁谱图进行拟合处理,两次拟合的结果不一致是什么原因?
在做方法时,我们通常为一个元素选多条谱线,但是拟合后,发现不同谱线浓度有时有很大的差异.大家说说有那些差异.
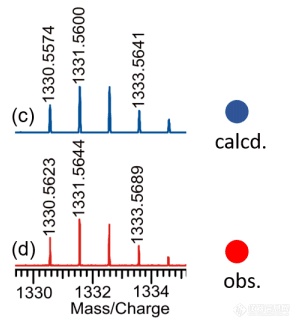
[color=#444444]我想拟合一个数据,大体上弄到如下图所示,想问下大神如何拟合质谱数据。[/color][color=#444444]谢谢![/color][color=#444444][img=,301,328]https://ng1.17img.cn/bbsfiles/images/2019/05/201905131540551793_4347_1847709_3.png!w301x328.jpg[/img][/color]
最近在做纤维素的固体核磁,需要对谱图中C4峰进行曲线拟合,得到纤维素的结晶信息,请高人指点拟合怎么操作啊?
前段时间做了一个纤维素C4区的NMR测试,对结果进行了分峰拟合。想请教各位大侠的是,如何将拟合出来的各谱线的基线弄到我用箭头表示的位置啊?就是让拟合出的谱线与得到的谱线的基线是同一个水平上的,谢谢![IMG]http://xa.photo.store.qq.com/http_imgload.cgi?/rurl4_b=42149b65270dcbb4207cf97f95bd0ed04c178b4b4a7077709389585df380c9e2fc131ef29ea15ff10005531b7169aa6033536ea3628a59e1784b9bc6f4ad494b44656cacafc15a77bfd1f1dcf58a440ed331b454[/IMG]
从事XPS的测试和解谱工作有一年左右,在这个论坛里也晃了有大半年的时间,虽然从未发贴,但帖子还是经常来看的.对于解谱,特别是峰的拟合还是有很多的疑惑,以前在论坛里讨论过,但总感觉没有讨论的太清楚,所以开贴将这个问题重新讨论讨论,希望前辈们能把自己的经验分享分享.我对峰拟合规则的认识如下:1.对于p、d、f等能级的次能级(如p3/2、p1/2,光电子能谱中一般省略/2,即为p3、p1)强度比是一定的,p3:p1=2:1;d5:d3=3:2,f7:f5=4:3。在峰拟合过程中要遵循该规则。如W4f中同一价态的W4f7和W4f5峰面积比应为4:3。2.对于有能级分裂的能级(p、d、f),分裂的两个轨道间的距离也基本上是固定的,如同一价态的W4f7和W4f5之间的距离为2.15eV左右,Si2p3和Si2p1差值为1.1eV左右。各元素能级分裂数据可参考网上数据库http://srdata.nist.gov/xps/Doublet_Sep.asp(这两条是我提供给学生的简单准则)对于FWHM(半峰宽)、L/G(洛仑兹和高斯的比例)等拟合时常见的参数有没有什么限制?
07年的时候去北京化学所刘芬老师实验室请教过拟合的问题,刘芬老师当初的解答主要有两点:1.扣背底选择平的点;2.半峰宽最大在2.0eV左右,峰宽与表面粗糙度有关。08年开表面分析会议的时候大连化物所盛世善老师讲到关于拟合的注意事项,提到:谱线的曲线拟合,应考虑:1,其合理的化学与物理意义2,合理的半高宽3,合理的G/L比:常取80%左右4,对双峰还应考虑:两个峰的合理间距、强度比、半高宽比*过渡金属的2p峰不对称,拟合较难此次讨论北京化工大学的程斌老师对于谱图拟合的见解:1、一定以物理意义为基础,而不是反过来;不要追求参差最小,而是追求物理意义;2、半峰宽以几个峰接近为好,3、合理的G/L比,对于我们新一代仪器,G可以100%,可以G/L80%4、对双峰还应考虑:两个峰的合理间距、强度比。从研究问题的角度程老师坚持第1点。必须以物理意义为基础来拟合,不追求参差最小。而不是以参差最小来确定物理意义。关于拟合的准则,大家的共同认识是相同的。具体的操作及拟合的心得需要多做,才有更多的体会。拟合本身具有极大的随意*,只要拟合符合上述准则,结合其他表征手段,能说明问题,能合理解释数据就ok。
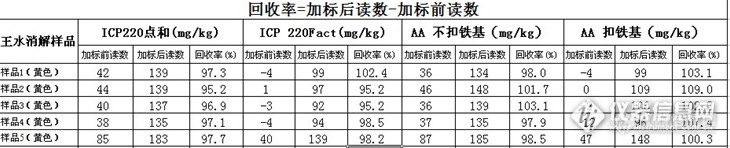
安捷伦(原瓦里安)FACT谱线拟合技术详解及可行性验证(以Fe对Pb的干扰数据分析)题外:论坛里有比较多的安捷伦(原瓦里安的用户),对光谱干扰造成的困扰讨论话题比较多,如Fe对Pb的光谱干扰,Pb的任一分析波长的峰形都不好,存在不同程度的干扰。含铁基类的样品在AAS上的Pb分析数据也是会有干扰值的,是故引出FACT谱线拟合技术来讨论如何消除谱线干扰(主要以Fe对Pb的干扰验证数据来分析)。其它元素的光谱干扰可举一反三。capidien版友写的【第二届网络原创大赛参赛作品】利用Varian FACT技术排除光谱干扰,对该技术做了详尽描述,本人不才,基于前代高手的基础上,利用实际研究的数据来论证安捷伦(原瓦里安)FACT谱线拟合技术的可行性。技术资料介绍:FACT,即Fast Automated Curve-Fitting,可以理解为“快速自动曲线拟合”。在干扰元素已知的情况下,这个功能可以很好的扣除干扰元素对待测元素的干扰。FACT 是对被分析元素和干扰元素的标样分别进行测量,用所得到的谱图数据进行图形解析,从而将被分析谱线旁边的干扰谱线分离出去。采用 FACT wizard, 你需要对被分析元素的纯标样、空白和干扰溶液进行测量。一旦你对每个模型的谱图感到满意后,该模型将被储存在方法中。然后可在该模型中测试 FACT 矫正的效果。在以后的分析当中,每条被测量谱线将依照其所建立的相应的模型对谱图进行解析。由被分析元素所产生的解析峰将参与被分析元素强度的计算。在一个方法中最多可建立多达 10 个 FACT 模型,在模型中可包括空白、被分析元素、基体和多达七个干扰物模型。基体和空白模型将对所有被分析元素有效。(该段背景介绍引用出处:capidien版友写的【第二届网络原创大赛参赛作品】利用Varian FACT技术排除光谱干扰)谱线拟合法首先通过建立ICP-AES谱图的数学模型,使校正ICP-AES分析中的光谱干扰问题转换为通过曲线拟合进行波形分解的问题,这实质上是用数学方法将重叠的总响应曲线分解成单个组分响应的分辨问题。金属样品的发射谱线多导致干扰多,背景高,考虑用安捷伦(原瓦里安)的Fact功能来校正。Fact的原理是用数学拟合的方法来将两条部分重叠的谱线还原成两条不同的谱线。Fe对Pb的光谱干扰,Pb的任一分析波长的峰形都不好,存在不同程度的干扰。以下几图为含有Fe基样品的Pb及Cd的峰形图:http://ng1.17img.cn/bbsfiles/images/2013/01/201301261902_422820_2329805_3.jpg图1.Fe干扰Pb在220.353波段的峰形http://ng1.17img.cn/bbsfiles/images/2013/01/201301261902_422821_2329805_3.jpg图2.Fe干扰Pb在217.000波段的峰形http://ng1.17img.cn/bbsfiles/images/2013/01/201301261903_422822_2329805_3.jpg图3.Fe干扰Cd在214.439波段的峰形 http://ng1.17img.cn/bbsfiles/images/2013/01/201301261903_422823_2329805_3.jpg图4.Fe干扰Cd在226.502波段的峰形[/font
各位坛友,我有个问题一直不太明白,就是在拟合Mo3d的XPS谱峰时的一些基本原则,现在已经明确的是对于确定的价态,比如Mo(IV),其3d3/2与3d5/2的面积比是2:3,半峰宽是1:1,峰间距是3.1-3.2,但是有个问题,Mo3d不同价态谱峰的半峰宽都是一样的么,比如Mo4+和Mo6+拟合谱峰的半峰宽难道都是一样的?还有一个问题,采用XPSpeak软件拟合的时候,其拟合曲线的高斯-洛仑兹的百分比是不是都必须分别保持一致?不知道我的问题说明白了没有,希望各位老师和坛友给予解答,谢谢!
维权声明:本文为gl19860312原创作品,本作者与仪器信息网是该作品合法使用者,该作品暂不对外授权转载。其他任何网站、组织、单位或个人等将该作品在本站以外的任何媒体任何形式出现均属侵权违法行为,我们将追究法律责任。 Origin 在化学化工实验数据处理中的应用 之基于Origin软件的多峰曲线拟合Origin 是美国OriginLab 公司(其前身为Microcal公司) 开发的图形可视化和数据分析处理软件,该软件主要体现2 大功能———数据分析和图形绘制。Origin 的数据分析主要包括数理统计、信号处理、图像处理、峰值分析和曲线拟合等各种完善的数学分析功能。准备好数据后, 进行数据分析时, 只需选择所要分析的数据, 然后再选择相应的菜单命令即可。Origin 的图形绘制则是基于模板, Origin 本身提供了几十种二维和三维绘图模板, 而且允许用户自己定制模板。绘图时, 只要选择所需要的模板就行。用户可以自定义数学函数、图形样式和绘图模板, 可以和各种数据库软件、办公软件、图像处理软件等方便的连接 。下面, 以化学化工实验数据为基础, 展示了Origin 在化学化工实验数据的图形控制中的一些具体做法。 Origin 是一款国际通用的具有超强功能的数据分析处理和科学绘图软件, 特别适合于绘制一些数据繁多计算复杂的图形。线性拟合 数据绘出散点图、点线图后,即可进行线性拟合:选择Analysis菜单下的Fit Linear或Tool s 菜单中的FitLinear ,即可对数据点进行线性拟合,在右下角的结果窗口中显示拟合直线的公式(斜率、截距值) ,以及相关系数和标准偏差等结果. 在线性拟合时,利用屏蔽工具(Maskpointtoggle ,屏蔽数据点) 、(Mask range ,屏蔽数据段) 可屏蔽某些偏差较大的数据点,以降低拟合直线的偏差.非线性(曲线) 拟合 Origin7. 0 提供了多种非线性拟合方式. 当数据绘出散点图或点线图后,可选择Analysis菜单下的Fit Polynomial (多项式拟合) 、FitExponential Decay (指数衰减拟合) 、FitExponential Crowt h (指数增长拟合) 、FitSigmoidal ( S 形拟合) 、Fit Gaussian ( Gaussian 拟合) 、及FitMulti- peaks (多峰拟合) . 在Tool s 菜单中提供了多项式拟合和S 型拟合,此外在Analysis菜单中的Non - Linear Curve Fit 选项Norr - Linear Curve Fit 选项可让用户自定义函数 .实验数据处理时,可根据数据图形的形状和趋势选择合适的拟合函数和参数,以达到最佳拟合效果. 多项式拟合适 用于多种曲线,且方便易行. 点击Analysis菜单中的Fit Plynomial 或Tool s 菜单中P
最小二乘曲线拟合在原子荧光光谱分析中应用摘 要: 曲线拟合是目前数据处理的基本方法之一,通过在微机上用Visual Basic语言编制程序,将实验数据使用最小二乘曲线拟合,构造一个最佳通用多项式拟合模型。再利用得到的函数关系实现对原子荧光光谱仪的实验数据进行多次曲线拟合与仿真,以扩大仪器的测量范围。
在做方法时,我们通常为一个元素选多条谱线,但是拟合后,发现不同谱线浓度有时有很大的差异.大家说说有那些差异?
用JADE分析出有两个物相,现在要计算其中一个物相的平均晶粒大小,在图谱拟合的时候是不是要把另一个物相的寻峰记录删掉再拟合?另外教程中说D值取1-2,那一般取多少呢?求高人指点~~~~
rietveld全谱图拟合做定量分析时,我用了标准物质来检验系统误差和方法的准确性,拟合结果还可以,各物相的含量和我配的物质含量比较吻合.可是RP 值和RWP值却很大,到了32%左右,峰形拟合的也不太好,不知是什么原因?
跟大家讨论一个棘手的问题,就是全普拟合前是否应该扣除 Kα2,大多数都认为不应该扣除,会使xrd数据失真,我也一直赞同这样的观点,最近又碰到同样的问题,就是扣除前和扣除之后峰形完全改变,这样直接导致了精修结果的变化,如下图,扣除前,39°附件主峰右边有个小峰,扣除之后,小峰就没有了,(扣除了 Kα2的缘故),因此,扣除前用单斜相精修的很好,而扣除后就应该用立方相精修了,我在黄继武老师编写的jade使用手册里面看到这样一句话“在精确计算点阵常数前必须将 Kα2 扣除”,和一位高手也讨论过,他说以前,是要求扣的,因为那时候人们很少用全谱拟合的方法,现在全谱拟合很普遍了,用全谱拟合时,不建议扣,因此我就茫然了,主要困惑以下几点:1.全普拟合前是否应该扣除Kα2;2.一般的精修软件(比如Fullprof,GSAS等)是否会扣除Kα2,或者有扣除Kα2的功能;3.扣除前和扣除后结果不一样,导致的原因在哪,哪一个结果更可靠;4.一般软件扣除Kα2背景的原理又是什么。种种困惑,希望大家不吝赐教!另外附上xrd的txt格式数据,以及扣除前后39°附件峰形的变化图







 我要推广仪器
我要推广仪器
 下载APP
下载APP