石墨炉测乳粉中铅和铬样品空白值很高 浓度都能到40ng/ml 不知道怎么回事 样品空白和我第四个标样浓度一样 样品值也很高 减去空白还可以有高人可以指点一下吗 另外铅的标准曲线也不好做995都不好做 大家做铅和铬都怎么处理样品 我是压力罐消解的
石墨炉做了六个样品以后,标准的吸收线突然变得像空白一样低,有谁碰到过类似的问题吗?[em63]
新买的耶拿650p石墨炉测痕金样品空白很高 求救啊仪器才装好,做了2次痕金,标准是0-40ug/ml ,但是样品空白做到了10ug/ml ,吸光度达到0.03,太高了 两次都是。标准曲线是0.998,水是超纯水,试剂应该没有问题,到底怎么回事啊,小女刚刚用,不懂啊!求各位大侠指教!
仪器是岛津AA6300C,用石墨炉测Cd,标准曲线点是0,0.5,1,2,4ppb,线性相关性是998,基改是1%磷酸氢二铵,测1%硝酸时吸光度是0.004,样品空白(两个平行)吸光度是0.2左右,实样浓度是1.5ppb左右,测了四川大米标准物质(平行)吸光度是0.3左右,实样浓度是2.3ppb,前处理用的是湿法消解。请问各位大神这种情况下样品空白高,可能存在哪些问题,怎样操作排查呢?新手急等,谢谢
先后用硝酸镁和磷酸二氢铵溶液来寻找测镉的最佳基体改进剂条件,在标准空白(1%硝酸)和标准溶液中加入0.5%和1%的磷酸二氢铵溶液发现仪器对镉的灵敏度增加了不少,但是标准空白加了磷酸二氢铵后,空白居然到了0.0900,1%的硝酸不加任何改进剂只有0.0080的吸光度,而做出的标准曲线截距很大。在0.0400-0.0500,我单独测了不同浓度的磷酸二氢铵,发现吸光度居然有0.0600左右,我的磷酸二氢铵是优级纯的,用的超纯水配制的,是不是磷酸二氢铵出了问题,我还对石墨炉升温中的灰化温度从350调到400,450,500,而原子化温度也从1700降低到1300,是不是还有升温程序的原因??
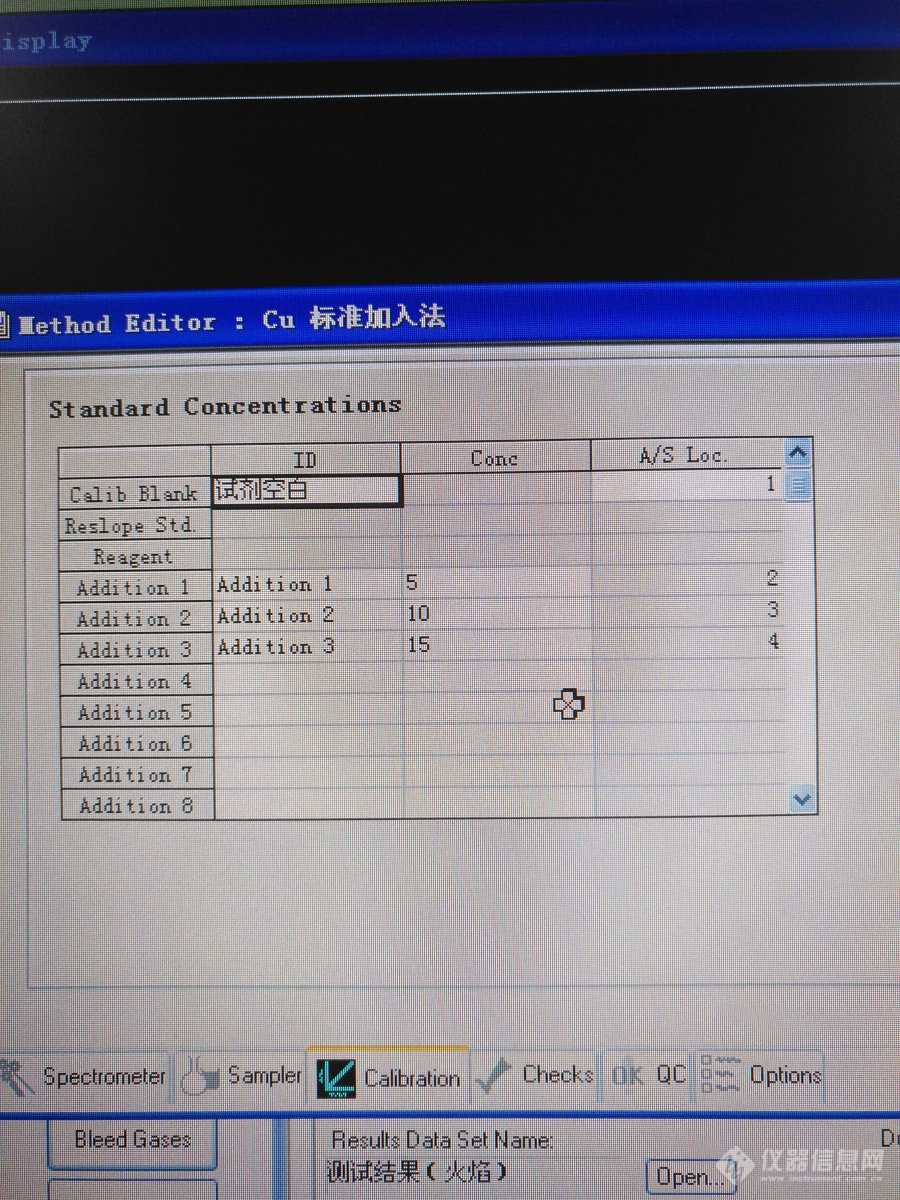
我用的是PE800。这里之所以单问石墨炉是因为火焰法做标加时直接把试剂空白当作标准空白将随后测得的信号减去这个值。但石墨炉标加法另有一套。它有自己考门的标准空白,并将随后测得的各信号中扣去这个空白值。标准空白测试试剂空白,然后依次测样品、添加一、添加二、添加三、添加四,出校准曲线同时给出样品的含量,但这个含量却没有扣除试剂空白:既没有扣去其信号值,也没有扣去其含量。怎么?难道要自己去扣除吗?如果是这样,该怎么扣?将样品的信号值扣去试剂空白?还是先求出试剂空白的含量?再说试剂空白的含量怎么求呢?当然这个倒容易,只是要有个说法,不能想咋咋吧?[img]http://ng1.17img.cn/bbsfiles/images/2018/03/201803261725426779_2474_2076515_3.jpg[/img][img]http://ng1.17img.cn/bbsfiles/images/2018/03/201803261726569727_3572_2076515_3.jpg[/img][img]http://ng1.17img.cn/bbsfiles/images/2018/03/201803261740295417_7040_2076515_3.jpg[/img][img]http://ng1.17img.cn/bbsfiles/images/2018/03/201803261741241157_1723_2076515_3.jpg[/img][img]http://ng1.17img.cn/bbsfiles/images/2018/03/201803261743006477_4749_2076515_3.jpg[/img]
最近一段时间用石墨炉测铅,试样空白的吸光值在0.03左右,样品的吸光值在0.004-0.009之间,这个是什么原因啊谢谢
如题石墨炉,用标准加入法测元素,需做试样空白吗?
石墨炉测铅的准确度经常受样品基体及样品空白的困扰,本人目前用北京化工厂的优级硝酸处理样品,样品空白浓度居然大于5ppb,请问大家都用什么酸处理样品?急需大家帮忙推荐好的优级硝酸和高氯酸。谢了!
用石墨炉测植物样品种铅时,试剂空白(硝酸:高氯酸=4:1)值较高,与灰化法的结果差异较大。不知道大家如何看待这个问题?
请教下各位 [url=https://insevent.instrument.com.cn/t/Wp][color=#3333ff]原子吸收[/color][/url]石墨炉法检测食品,标准和样品的空白值一般为多少算合格?样品空白一定要比标准空白高吗?样品空白用1%的硝酸定容,标准空白为1%的稀硝酸。谢谢!
[url=https://insevent.instrument.com.cn/t/Wp][color=#3333ff]原子吸收[/color][/url]石墨炉法测水中的镉,根据需要点击空白校正,工作站生成如下表 浓度 吸光度 空白校正标准系列浓度1 标准系列浓度2标准系列浓度3标准系列浓度4标准系列浓度5标准系列浓度6空白校正样品1样品2我知道样品溶液的吸光度减去空白溶液的吸光度,但是标准系列浓度的吸光度是否也要减去空白溶液的吸光度?

(1)仪器型号:AA-7003A(2)背景扣除方式:氘灯(90)(3)分析模式:石墨炉(4)待测元素名称:Sn(5)标准样品浓度:0ug/L(2%硝酸)(6)检查过程:打入20uL的硝酸进入石墨炉测量,吸光度很高(7)测试结果和图谱:测量过几次有0.0456、0.1256、0.0756、0.1554等等,所用的石墨管是新的,测定Cd,打入同样的量跟试剂,所测吸光度是0.0120、0.0101。如图是测量的条件,还有吸光度的走向图(8)疑问和求助内容:换了一次仪器内部的通水管路之后,测量Sn这个元素的空白吸光度就变得很高,测量Cd、Pb这两个元素倒没有很高。在换之前测所有的元素空白值都挺正常的,换了之后就唯独Sn这个元素的空白吸光度变得很高(前后测Sn所用参数一致)或许是因为灯的原因?Sn元素灯坏了?[img=,690,264]https://ng1.17img.cn/bbsfiles/images/2019/09/201909021050584855_1011_3983958_3.png!w690x264.jpg[/img][img=,690,592]https://ng1.17img.cn/bbsfiles/images/2019/09/201909021051087790_4193_3983958_3.png!w690x592.jpg[/img][img=,690,517]https://ng1.17img.cn/bbsfiles/images/2019/09/201909021051214750_2464_3983958_3.jpg!w690x517.jpg[/img]
请教各位大神,我用的仪器是Thermo ice3500,测试时,高浓度的标准液吸光度低于低浓度标准液,标准曲线不成线性,显示MD180,校正失败,无法计算试样浓度是怎么回事?测试设置参数如下:光谱仪参数 元素: Cd 测定模式: 吸收 波长: 228.8 通带: 0.5nm 灯电流:% 50 背景校正: 塞曼 高速采集信号: 关 最佳化光谱仪参数: 否 信号类型: 瞬间 瞬间信号类型: 高度 重测试样数目: 3 测量时间:秒 3.0 剔除模式: 否 测定峰值从: 0.00 到: 3.00秒 使用RSD%测试: 否 石墨炉参数 石墨管型号: 普通 注射温度:℃ 0 石墨炉程序 (秒) 78.0 阶段 温度 时间 斜坡 气体 气体 命令 (℃) (秒) (℃/秒) 类型 流量 1 90 20.0 10 2 0.2L/min 2 100 20.0 10 2 0.2L/min 3 400 20.0 150 2 0.2L/min 4 1100 3.0 0 2 关 RD TC 5 2500 3.0 0 2 0.2L/min TC 进样参数 进样: 石墨炉 自动掺液准备: 否 工作体积:ul 20.0 进样体积:ul 20.0 改进剂 方法 体积 顺序 1 无 20.0 1 2 无 20.0 2 3 无 20.0 3 4 无 20.0 4 5 无 20.0 5 6 无 20.0 6 试样制备: 稀释 标准准备: 固定 标准加入: 无 慢速溶液提取: 否 慢注射: 否 进样延时: 否 清洗次数: 1校正参数 校正模式: 普通 线性拟合: 线性 使用已存储的校正曲线: 否 浓度单位: ug/L 比例单位: ug/L 比例因子: 1.0000 可接受的拟合: 0.995 重新调整限制:% 10.0 失败动作: 标记并继续 主标准 2.0000 标准1 0.2000 标准2 0.4000 标准3 0.6000 标准4 0.8000 标准5 1.0000QC 参数 测试 浓度:ug/L 限制 动作 重复 QC空白 0.0000 -0.0100 到 0.0100Abs (峰高) 继续 继续下一个元素 QC检查 1 1.0000 90.0 到 110.0% 继续 继续下一个元素 QC检查 2 1.0000 90.0 到 110.0% 继续 继续下一个元素 QC检查 3 1.0000 90.0 到 110.0% 继续 继续下一个元素 QC检查 4 1.0000 90.0 到 110.0% 继续 继续下一个元素 QC检查 5 1.0000 90.0 到 110.0% 继续 继续下一个元素 QC检查 6 1.0000 90.0 到 110.0% 继续 继续下一个元素 QC检查 7 1.0000 90.0 到 110.0% 继续 继续下一个元素 QC检查 8 1.0000 90.0 到 110.0% 继续 继续下一个元素 QC检查 9 1.0000 90.0 到 110.0% 继续 继续下一个元素 QC制备掺液 1.0000 90.0 到 110.0% 继续 继续下一个元素 QC分析掺液 1.0000 90.0 到 110.0% 继续 继续下一个元素 QC稀释 1.0000 10.0% 继续 继续下一个元素 QC校正空白 -1.0000 到 1.0000ug/L 继续 继续下一个元素QC TEST SUMMARY QC ACTION QC SUMMARY RESULT 无QC动作执行测试结果是下面这样的,高浓度的标准液吸光度低于低浓度标准液,标准曲线不成线性,显示MD180,校正失败,无法计算试样浓度是怎么回事?RESULTS FOR ELEMENT Cd SAMPLE ID RESULT TYPE SIGNAL 背景 Rsd FLAGS CONC. CORRECTED CONC. TIME DATE Abs (峰高) % ug/L ug/L 空白 平均值 0.0018 33.2 0.0000 空白 重测试样3 之 1 0.0020 -0.0009 19:48:58 2019/6/10 空白 重测试样3 之 2 0.0011 -0.0010 19:50:58 2019/6/10 空白 重测试样3 之 3 0.0022 -0.0013 19:52:58 2019/6/10 标准1 平均值 0.0574 3.0 0.2000 标准1 重测试样3 之 1 0.0557 0.0049 19:54:58 2019/6/10 标准1 重测试样3 之 2 0.0574 0.0050 19:56:58 2019/6/10 标准1 重测试样3 之 3 0.0592 0.0051 19:58:59 2019/6/10 标准2 平均值 0.0558 0.7 0.4000 标准2 重测试样3 之 1 0.0560 0.0044 20:00:59 2019/6/10 标准2 重测试样3 之 2 0.0560 0.0043 20:03:00 2019/6/10 标准2 重测试样3 之 3 0.0553 0.0048 20:05:00 2019/6/10 标准3 平均值 0.0480 0.8 0.6000 标准3 重测试样3 之 1 0.0483 0.0041 20:07:00 2019/6/10 标准3 重测试样3 之 2 0.0481 0.0041 20:09:01 2019/6/10 标准3 重测试样3 之 3 0.0476 0.0049 20:11:01 2019/6/10 标准4 平均值 0.0457 0.8 0.8000 标准4 重测试样3 之 1 0.0453 0.0031 20:13:02 2019/6/10 标准4 重测试样3 之 2 0.0459 0.0035 20:15:02 2019/6/10 标准4 重测试样3 之 3 0.0460 0.0034 20:17:02 2019/6/10 标准5 平均值 0.0417 4.4 1.0000 标准5 重测试样3 之 1 0.0396 0.0033 20:19:02 2019/6/10 标准5 重测试样3 之 2 0.0429 0.0032 20:21:03 2019/6/10 标准5 重测试样3 之 3 0.0426 0.0033 20:23:03 2019/6/10 试样空白 平均值 0.0012 8.3 试样空白 重测试样3 之 1 0.0011 -0.0005 20:25:03 2019/6/10 试样空白 重测试样3 之 2 0.0013 -0.0007 20:27:02 2019/6/10 试样空白 重测试样3 之 3 0.0012 -0.0015试样标识 1 平均值 0.0037 24.8 试样标识 1 重测试样3 之 1 0.0029 1.1147 20:49:05 2019/6/10 试样标识 1 重测试样3 之 2 0.0035 1.0817 20:51:05 2019/6/10 试样标识 1 重测试样3 之 3 0.0047 0.8988 20:53:05 2019/6/10 试样标识 2 平均值 0.0120 52.4 试样标识 2 重测试样3 之 1 0.0108 1.6042 20:55:04 2019/6/10 试样标识 2 重测试样3 之 2 0.0188 1.7657 20:57:04 2019/6/10 试样标识 2 重测试样3 之 3 0.0064 1.3034 20:59:03 2019/6/10 试样标识 3 平均值 0.0106 59.3 试样标识 3 重测试样3 之 1 0.0179 1.7369 21:01:02 2019/6/10 试样标识 3 重测试样3 之 2 0.0073 1.4560 21:03:03 2019/6/10 试样标识 3 重测试样3 之 3 0.0067 0.9291 21:05:03 2019/6/10 试样标识 4 平均值 0.0024 71.5 试样标识 4 重测试样3 之 1 0.0011 0.8412 21:07:03 2019/6/10 试样标识 4 重测试样3 之 2 0.0018 1.1386 21:09:02 2019/6/10 试样标识 4 重测试样3 之 3 0.0044 0.8336 21:11:02 2019/6/10
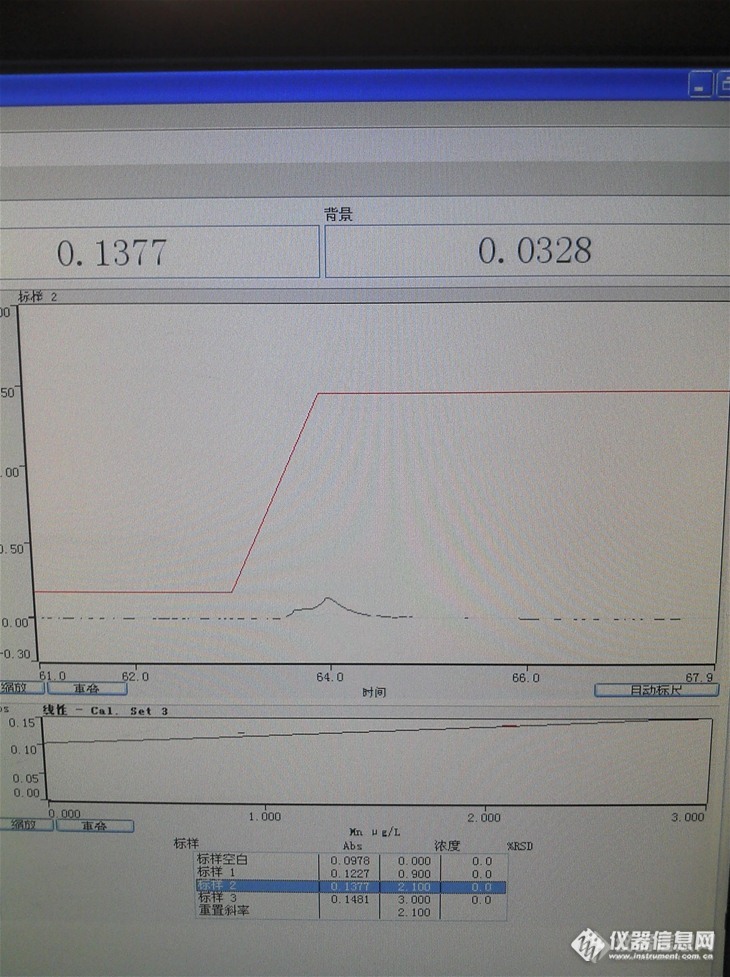
仪器型号:瓦里安240Z待测元素名称:锰分析模式:石墨炉背景扣除方式:塞曼标准样品浓度:0.9、2.1、3.0 ug/L故障现象:出峰异常、峰形不好、空白值高http://ng1.17img.cn/bbsfiles/images/2013/04/201304111656_434982_2665078_3.jpg检查过程:做其它项目的时候空白很低的,纯水应该没有问题疑问和求助内容:我是第一次用石墨炉做锰,灰化温度650、原子化温度2300
仪器条件:波长283.3 狭缝0.7 线性记算截距,基改到用的磷酸二氢胺-销酸钯,进行背景校正,标样浓度最大20ppb,标准梯度自动稀释,标准曲线有0.9991。样品处理按国标加的浓销酸微波消解,150度2小时赶酸后用千分之三销酸定容至25毫升上机检测。上机后经常出现试剂空白铅约4ppb,而样品则约-4ppb(减试剂空白后),回收率做的100PPb的样品约75%.想请教下除了以下原因还有没有其它原因。谢谢!1.器皿污染,未用销酸泡干净。2.玻璃器皿本身含铅,3.其它如扣背景,换波长,狭缝,调整石墨管升温经程序的读数时间等,想问问各位老师最可能的问题是什么。
最近在做甘草中铅的检查,用的是石墨炉法,样品处理时干法消解,取样0.5g定容至25ml。铅标准液的浓度分别为0ng,5ng,20ng,40ng,60ng.加入了基体改进剂之后,测量的数值居然是空白为正值,样品和铅标准液都是负值,期盼各位大侠给支个招,如何解决?
石墨炉检测铅一般样品空白吸收值是多少正常?我8ml硝酸加1ml双氧水微波消解后定容25ml吸收值0.0423正常吧
RT。。之前还以为是旧石墨管,才出现空白高。但换了新的结果还是一样,可是我做的标准物质测出来却在质量控制范围内,样品有部分时候会比空白低。。这样的结果到底可不可信呢??(空白吸收值0.024,浓度8.0ppb) 谁可以提供某个浓度标液的参考吸收值呢?用 的岛津AA-7000的仪器。。。感激不尽啦
石墨炉法测钙标准溶液2.5ppb扣空白后有时候吸光度0.17,有时候0.15是怎么回事?
石墨炉测铅为什么样品空白有浓度?什么原因?
我用石墨炉做铝,用热解石墨管,现在发现空白值很高,水的吸光值有0.1左右,可是样品空白更高,样品空白的吸光值有0.8-0.9这样,请教高手,做铝时有哪些需要注意的,或者有什么改进的方法吗?
仪器型号:岛津AA-7000待测元素名称:Pb分析模式:石墨炉背景扣除方式:氘灯标准样品浓度:1、2、5、10、20ppb未知样品的处理方法:微波消解(0.2g样品用8ml硝酸和2ml过氧化氢溶解,130℃保持10min,150℃保持5min,180℃保持10min)故障现象:进样时发现样品吸收值是负值,标曲有0.9996,吸收值也正常疑问和求助内容:怎么会出现负值?我是用3%的硝酸溶液作为空白的,结果样品空白和样品吸收值都显示负值,郁闷。
石墨炉测铅时,样品空白大概多少?我用加硝酸5ml微波消解,25ml定容,结果空白值好像也挺高的大概10ug/L,做了很多次也是这样的数值,这是正常呢,还是有其他原因引起的?消解罐?硝酸用的是默克的产品。请各位老师给点意见,还有你们的经验告知,谢谢!!
新接触PE AA800,用石墨炉做铅标准曲线,相关系数3个9,但做样品时吸光度(或测出铅浓度)总是为负值,数据显示样品含铅量低于样品空白,重处理后值可为正,但还是低于样品空白,求教原因?
请教一下各位:原子吸收石墨炉:做标准空白(1%的硝酸)测Pb时的吸光度一般为多少算合格?我这几天测出来是0.14,0.08,0.18,可以吗?
石墨炉原子吸收测植物蛋白饮料中铅含量时空白特别高,干法消解,以前没有过此情况。纯净水的吸光值为0.09,但是测得5ng/ml的铅标准使用溶液的吸光值为0.089,测样品时背景特别高,不知道怎么回事,希望有经验的老师知道!!谢谢
PEAA800,砷标,用超纯水做标准空白,出现负值,大概在-0.0020左右,而且非常稳定,进空气的值也差不多是这个值,使用的是环状石墨管,如果使用平台式的石墨管标准空白就恢复正常了,接近0。 做标准空白时候的吸收线都在0点以下波动,现在想不明白原因,但是这个标准空白负值对测定倒是没有影响,因为十分稳定,而且做过茶叶标准物质,测量值仍是梃准。大家讨论一下啊,这种现象给人一种似乎吸收线整体往下偏移了,或者说0点基线整体往上提了的感觉。
鉴于还没收到版友提出的问题,现自选主题先发一贴,水平有限,勿笑。扫盲系列活动,以后尽量每周末更新一贴,下周末将由haodou000版主来发贴。欢迎版友提出问题,大家一起学习、总结和归纳。AAS测样品空白值高原因有两大类:一、留有残余,造成记忆效应。1、最常见原因是刚做完标准曲线马上就测空白试剂,特别是标准曲线浓度较高的情况下,造成记忆效应,空白值过高。分述如下:2、火焰法空白值过高,进纯水或空白试剂清洗一段时间再测。3、石墨炉法空白值过高,高温空烧石墨管1~3次。4、因石墨管积炭过多而空白值高,通过通空气去除多余积炭,对石墨管寿命有影响,慎用。PE的仪器有一路气体是空气(跟火焰法的空气管道不是同一条),用钢瓶的压缩空气在净化阶段通入少量空气(通氩气同时同很少的空气),或者在净化结束后通入空气运行灰化、原子化、净化,具体参数根据样品而定。注:此条(一类第4条)由haodou000版主提供。5、石墨管寿命已到期,更换管子。6、氢化物发生器石墨炉法空白值过高,因还原剂(硼氢化钠或硼氢化钾)配制而引起,重新配制,还原剂需现配现用;因石墨管而引起,高温空烧管子1~3次;因氢化物发生器而引起,调节泵速、Ar流量,检查通氢化物的进样管是否有液体,若有则需吹扫干净。二、前处理带来的污染。污染源有3:1、实验室用水的污染 2、酸或试剂本底较高 4、消解过程带入的污染首先判断。举例:对于石墨炉法,先空测(即不进任何样品,让自动进样针扎没放样品的地方),若正常(响应值不大)则一般可排除不是石墨管残留的问题,否则高温空烧石墨管;再进纯水,若也正常,则可证实不是实验用水的问题,否则须换实验室用水;若前面两步都没问题,至此就可判断是酸本底的问题或是消解方法的问题,对于酸本底高则换酸,若换了本底较低的酸,样品空白值还是很高的话,则表明是消解方法的问题,须改善或更换消解方法。对于火焰法和氢化物发生器石墨炉请参照上面的思路,不再阐述。接着仔细审视整个前处理过程。对于消解过程带入的污染是可以是很多方面:容器器皿清洗不干净、仪器设备带入的污染、玻璃容器金属离子的析出、通风橱灰尘的掉落、环境因素等。这一过程要具体问题具体分析,举例说明:测奶粉中的9种矿物质元素,我用干法灰化,过程如下:首先在电炉上加热炭化至无烟,此时样品呈灰白色,接着转至马氟炉灰化至亮白色,最后取出再加酸溶解定容。最后测得的结果是样品空白的铁较大(我用PE的ICP测的,仅以此说明消解方法引入的污染),而且样品中的铁也超标。后来经多次寻找原因,发现是在马氟炉灰化时出的问题,那个马氟炉已用多年,里面已有铁锈。。。。。。后来新买了个马氟炉,问题也就解决。仅以此抛砖引玉,如果排除是仪器的原因,则要在前处理过程的每个步骤寻找原因。haodou000版主建议:实验前,冲洗几遍进样针、石墨管空烧两次(这是个很好的习惯,本人每次测样前都烧一次)。测Na、K等要注意自来水的污染,器皿应用超纯水冲洗。水平有限,有错漏之处请各版友指出(积分奖励),欢迎发贴讨论、补充。感谢haodou000版主提供的帮助!本版专家“开心”补充: 1、 如果楼主指出的这几点都排除了,空白吸光度值还是高,那就是基线飘了。在做火焰AA时,在第一次测空白时,吸光度值如果很大时,最好再测一次。当空白的吸光度值的三次读数均为0.000或-0.000时,再分析校准曲线,同理,当发现做样过程中数据有波动了,就再分析一次空白。如果空白的吸光度值不为0,就再测一次直到吸光度值归零。原理有些类似分光光度计的参比池调零。 2、 如果是石墨炉的空白很高,排除溶液,容器,进样针等污染因素外。可以检查一下是否是石墨管装反了。平台石墨管的平台联结点应朝里放置,不可朝外。感谢开心~
[em04] 最近突然发现做石墨炉测铅时空白样品吸光度很高,居然达到0.08-0.10,但是曲线的线性还是可以的,基本相关系数达到0.99以上,一直试了很多方法,仍然解决不了,包括试验过高纯水、硝酸、比色管等的空白,发现都不是主要的影响因素,最后当我黔驴技穷时,打开实验室所有窗户通风,并把石墨炉加热程序中的原子化温度和清除温度相应提高了300度,空烧了三次,重新测空白时发现,一切都正常了,空白值为0.009-0.015,究其原因:一是环境中铅污染,如果是这样的话,以后各位实验工作者可是要注意实验室通风,保护自己的身体啊,毕竟如果造成重金属中毒那可是对不住自己啊;二是石墨管的记忆效应太大引起空白高,通过提高原子化温度和清除温度可以得到改善。 以上一点粗浅经验,贴出与各位共享,希望各位多交流自己在测石墨炉时的经验之谈,以达到共同提高的目的。







 我要推广仪器
我要推广仪器
 下载APP
下载APP