用石墨炉测食品添加剂中的铅有没有标准?
今天一个人在单位忙活了一下午 尝试了一下石墨炉直接进样标准加入法测奶粉铅奶粉为脱脂奶粉质控品 铅0.03-0.018mg/kg 取3g定容到25ml溶解10ul进样量+10ul稀释液+5ul硝酸钯配置铅标准10ug/l 加标曲线2 4 6 8ug/l灰化温度450 20秒 800 20秒原子化温度1600结果不是很好看 呵呵呵 线性极差 舍去两点可以成线性 质控值在范围内信号很低 空白信号0.0005 样品信号0.0064 样品+2ug/l信号才0.0086 样品+8ug/l信号才0.0150做完后石墨管内积碳很多(没办法 没有接special空气)讨论1.第一次做标准加入法 线性很不理想 才一个9 不知道是加标系列不合理 还是加标量太小 记得大家说第一点最好和样品接近 2.我配置的铅标准没有加硝酸 纯水配的 怕奶粉沾了结块 3.不知道纯水配的铅和1%硝酸配的铅信号有没有区别 纯水配置能保持几天4.昨天配的铅标准用的1%硝酸 放置在一种半透明的软的塑料的咳嗽药水瓶里 今天测下来10ug/l尽然信号到了0.0708 昨天0.0556 这是怎么回事
[求助]马弗炉高温消化石墨炉法测茶叶中的铅,含量比标准值差很多!称1g茶叶,马弗炉中500度消化6h,2ml0.5mol/L硝酸溶解,然后洗入25ml比色管定容。上石墨炉。4.4(+-0.3)ppm的茶叶质控样,测量后计算得到的结果只有0.38ppm。难道是马弗炉500度太高,把铅都挥发了?
最近在做甘草中铅的检查,用的是石墨炉法,样品处理时干法消解,取样0.5g定容至25ml。铅标准液的浓度分别为0ng,5ng,20ng,40ng,60ng.加入了基体改进剂之后,测量的数值居然是空白为正值,样品和铅标准液都是负值,期盼各位大侠给支个招,如何解决?
求助:石墨炉[url=https://insevent.instrument.com.cn/t/Wp][color=#3333ff]原子吸收[/color][/url]做镉,铅,铬时。为何回测标准不好。 标准曲线还好。只是要做好几次才能做好。而我把稀释的标准放到样品栏去打,就打不出来。标准浓度在线性范围内。 还请高手指教!!! 急。。。[em0808]
最近在摸索AA6300石墨炉方法,因为是新接手的仪器(好几年都没有人用过这个仪器了),前几天刚刚通过了检定。领导说做铅先试试。于是就先做了铅的曲线和质控物质10052茶叶。质控物质的铅含量在1.4~1.8mg/kg范围内。 前天进行的前处理,使用的是干灰化法消化。小火在电炉上炭化至无烟后,放入马弗炉500度灼烧6小时。取出后再加入1ml(1+1)HCL在电热板上130度溶解残渣,后用2%(体积比)HCL定容25ml待测。 前天先用原子荧光测了一下,最后铅的结果是可以满足质控物质的范围的。昨天用原吸做了一下,结果浓度值只是范围的四分之一。很是不解。因为要是前处理有问题的话,那在原子荧光上面出来的浓度值也有问题才对。 我的原吸条件如下: 石墨管:高密石墨管, 氘灯扣背景。http://ng1.17img.cn/bbsfiles/images/2012/03/201203102001_353770_1766615_3.gif ABS=0.010192Conc+0.0016508 r=0.9994测定的标准曲线的吸光度在仪器手册给定的值附近,但是测定质控物质时候,吸光度一下子就下去了,而且背景线很高,将样品峰包裹在里面。样品峰也就是背景峰的一般。是不是没有加基体改进剂的问题?加什么基改效果更好。加多大浓度的呢?
大家用石磨炉法测铅的检出限是不是用 3×标准偏差/斜率 来算?我作的标准曲线斜率是0.00233,其它数字除以它不是会变得很大吗?晕倒我用消解食品的混合酸作空白,20组,标准偏差0.05,这样一算检出限=64ppb [em06] [em06] [em06] [em06] [em06] [em06] [em06] [em06] [em06] [em06] 而且还没计算稀释倍数,要是算进去的话什么都别想测了???大家用石磨炉法测铅的检出限大概是多少?
我使用的是瓦里安的AA240石墨炉,在测标准曲线过程中,发现同一浓度标液两次吸光度值相差很大,请问哪位能告诉我怎样解决一下这个问题!
用石墨炉法测铅元素,加的基体改进剂是磷酸二氢铵-硝酸钯,完全根据标准5009.12-2017,标准曲线相关系数才不到0.99,不加有0.995以上,是否可以不加,样品是测大米。或者是否是基体改进剂的纯度不够
用石墨炉测定铅、镉的标准系列是多少?
用石墨炉测定生活饮用水中铅,标准系列有问题请教: 为什么铅标准系列浓度越高吸光度越低? 石墨炉加热程序按照国标设定
我用石墨炉测铅,吸光度值太低,20PPb的进20uL吸光度才0.05多。不知道什么原因,我的标准溶液是用分析纯的硝酸铅配的,没用酸溶解,直接用二次水溶解了之后逐级稀释的,会有影响吗?还有就是测定时溶液中要有酸吗?要是需要,什么酸合适,酸的浓度又该是多少?我刚开始用石墨炉,好多问题呀?希望各位高手尽快给予小妹一点帮助,万分感谢!
刚刚使用石墨炉不久,这几天在建立检测血铅的方法。用的是Thermo S2石墨炉,氘灯扣背景,基改曲哪通。标准曲线是手工稀释高浓度标准溶液得到:0、5ug/l、10ug/l、20ug/l四个点,5ug/l的吸光度达到0.040,标准曲线拟合度99.8%的很好。但是在检测健康人样本时,结果全部是超标的,污染的可能性基本排除,请大家帮我想想办法,会是什么原因导致的?
石墨炉测牛血铅 标准曲线和前处理中加的血 是不是加牛血还是牛血铅的标准物质?
刚接触[url=https://insevent.instrument.com.cn/t/Wp][color=#3333ff]原子吸收光谱[/color][/url]新手,求教一下石墨炉测血铅的问题1、加入血样灰化时,有刺鼻烧焦头发样味,正常吗?2、如何判断石墨管报废了?3、能否贴一下石墨炉测血铅的具体做法?网上只搜到一些标准,找不到具体操作步骤。谢谢
最近石墨炉做标准曲线时出现了很反常的现象,五点标准曲线做出来线性差的不得了,换了新的石墨炉没有明显的改善之前测铅线性都能达到三个9以上很是苦恼,现配标准液也是不行,目前采用的是在线稀释很差自己配五级标液同样很差各位能否为小弟指点迷津!设备是PE600
各位前辈,最近两天一直用石墨炉做铅的标准曲线,过零点非线性方程,不加集体改进剂,条件如下step1,100°,坡升5秒,停留20秒;step2,140°,坡升15秒,停留15秒;step3,700°,坡升10秒,停留20秒,ste,4,1800°,坡升0秒,停留5秒;step5,2200°,坡,1秒,停留5秒。出现的问题如下:1,重复进样两针,偏差有时候很小2.0%左右,有时候就很大30%左右2,线性很差,除掉第一点的话有时候能到三个93,以2.5ppb的浓度进样,吸光率在0.012左右,不知道这个吸光率是不是正常的4,升温步骤几乎每次都要在石墨管那里冒烟以上已经困扰了我两天,更换过新的石墨管,调整进样针位置很多次。望各位前辈指点一二,不胜感激。补充:灯能量58-60,电流10A,狭缝0.7。
大家平时用石墨炉测铅的时候,测量模式中选“峰高“还是”峰面积“呢?两者有什么不一样呢?最近我发现用“峰高”检测出来的标准系列的吸光值比用“峰面积”检测的吸光值高好多,而且检测复杂组分的样品时,测出来的值就差了好多倍呢!
用石墨炉测植物样品种铅时,试剂空白(硝酸:高氯酸=4:1)值较高,与灰化法的结果差异较大。不知道大家如何看待这个问题?
求石墨炉测水中铍的标准曲线,第一次做不知道做的对不对,哪位能发给我标准曲线看一下。。
用[url=https://insevent.instrument.com.cn/t/Wp][color=#3333ff]原子吸收[/color][/url]石墨炉法测定铅标准溶液,浓度分别为5,20,40,60,80ng/ml,线性不好是什么原因?原子化温度是1800,灯电流20能量87[img]http://ng1.17img.cn/bbsfiles/images/2018/06/201806081639195014_1667_3423552_3.jpeg[/img][img]http://ng1.17img.cn/bbsfiles/images/2018/06/201806081639274339_5296_3423552_3.jpeg[/img]
石墨炉测铅污染了,2%酸水下来0.004,基改硝酸钯测下来0.03,单独测都好着,可是一混合,标准空白就0.05了,玻璃瓶,进样杯都换新酸泡过了,石墨管也换新了,灯也换了,石墨锥和石墨帽拆下来用乙醇清洁了,就是空白值下不来0.05左右,酸用优级纯和电子级都比较了,酸正常,石墨炉空烧0.003,这是什么原因,岛津AA6880
仪器型号耶拿700,石墨炉测铅,做标准曲线梯度5,10,20,30,40;想问下各位大佬的吸光度都有多少啊?我做的吸光度老是太低了,所以想问下大家都是如何解决的?学习学习
[size=4]各位老师好: 我是新手,刚接受一台新perkinelmer 的900Z单石墨炉[url=https://insevent.instrument.com.cn/t/Wp][color=#3333ff]原子吸收[/color][/url]分光光度计,以后主要用于尿铅、尿锰及血铅检测,有以下问题请教: 1.大家觉得尿铅、尿锰及血铅检测中分别用什么基改好(仪器推荐/国标/其它)?不用基改效果差别大不大?[/size][size=4] 2.具体的升温程序是怎样设定的?用仪器推荐吗?不同品牌型号的仪器测定同种物质的升温程序大不大?[/size][size=4] 3.峰形和标准曲线的线性哪个对检测准确度的影响大?如果线性较好而峰形不好时能否忽略峰形的影响?[/size][size=4] 4.血样的粘稠度较大,大家一般采用多少倍稀释后测定?有时候测完后在石墨管中留有泡沫样残渣是否为灰化不完全?如何解决?[/size][size=4] 以上是几个入门问题,还有更多疑问需要大家帮助解决,只能一步步慢慢来了,期盼大家能倾囊相授。谢谢![/size]
按照GB/T5750.6石墨炉[url=https://insevent.instrument.com.cn/t/Wp][color=#3333ff]原子吸收[/color][/url]分光光度法测水中铅,标准的吸光度变化不大,石墨管是新换的。又没有同样情况的,知道是什么原因吗?[img]https://ng1.17img.cn/bbsfiles/images/2021/09/202109061200138119_2973_5055194_3.png[/img][img]https://ng1.17img.cn/bbsfiles/images/2021/09/202109061200138011_8753_5055194_3.png[/img][img]https://ng1.17img.cn/bbsfiles/images/2021/09/202109061200137448_7499_5055194_3.png[/img]
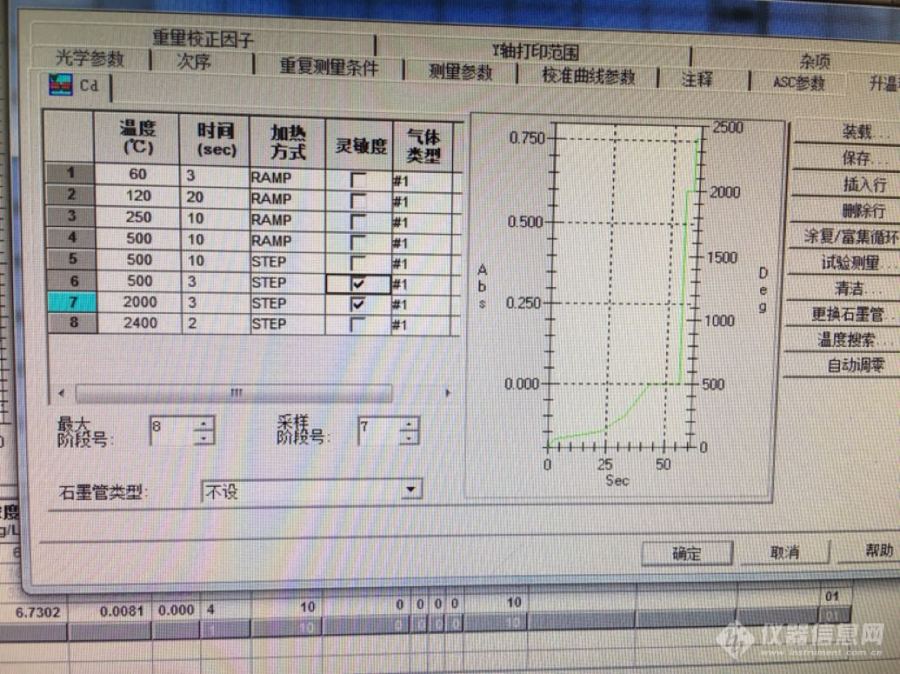
我用岛津AA6880 去年刚买的,石墨管今天刚换的已做过老化,石墨炉测土壤中的镉铅,标准曲线浓度按国标配的,自动进样做一遍曲线不行,手工配的曲线还是不行,空白吸光度0.0011,测量条件如下图,请前辈们指点指点问题出在哪里[img=,690,516]http://ng1.17img.cn/bbsfiles/images/2018/04/201804241237569053_2425_3392048_3.png[/img][img=,690,516]http://ng1.17img.cn/bbsfiles/images/2018/04/201804241237577371_5231_3392048_3.png[/img]
我用石墨炉做血铅标准曲线时,用自动加样器加标准和稀释液,共进样10微升,另外再加基体改进剂10微升,但是正常人的血样什么时候加?没有好的解决方法
今天用石墨炉测铅时,标准曲线一直做不好,用的是自动配制,铅的母液浓度为10ppb,浓度顺序依次为0,2,4,6,8,10,做出来空白的吸光度有0.0058,其余的点的吸光度都很小,斜率很低,基本不呈线性,做了两次都是如此。用了磷酸氢二铵做基改剂,进样体积10微升,基改剂体积也为10微升。条件:干燥温度为80,95,120;灰化温度为350,原子化温度为2100各位有没有遇到过这样的问题啊?会是什么原因呢?大家晒一晒讨论讨论吧^_^盼高手指教~
石墨炉测乳粉中铅和铬样品空白值很高 浓度都能到40ng/ml 不知道怎么回事 样品空白和我第四个标样浓度一样 样品值也很高 减去空白还可以有高人可以指点一下吗 另外铅的标准曲线也不好做995都不好做 大家做铅和铬都怎么处理样品 我是压力罐消解的
今天用石墨炉测钙片中的铅,用的是标准加入法,AAS240z仪器,铅的最高浓度是20微克/L,用的是自动配制,基体改进剂是20%的磷酸氢二铵,怎么标准曲线一直做不出来?基本上都呈先上升后平齐的趋势。会不会是基体太复杂了?那要怎么样才能消除干扰呢?(用标准曲线法的话,曲线就能做出来。)现在还是想考虑标准加入法,如何处理呢?谢谢啦







 我要推广仪器
我要推广仪器
 下载APP
下载APP