在做乳制品维生素A含量检测时发现新国标中色谱条件里柱温是20℃,之前旧国标里是35℃,想询问一下大家,柱温对含量检测的影响,谢谢
ASTM F2695-12方法检测维生素E含量,样品是混合塑料棒材
维生素C又叫抗坏血酸(Ascorbicid),广泛存在于植物组织中,新鲜的水果、蔬菜中含量较多。是一种水溶性小分子生物活性物质,也是人体需要量最大的一种维生素。维生素C具有还原性(其结构式如图1),可以与许多氧化剂发生氧化还原反应,因此可以利用其还原性测定维生素C的含量。目前食品中测定维生素C含量的方法主要有碘量法,是利用维生素C的氧化还原性为基础的一种氧化还原方法。冈其酸度不易把握,碘需要标定且易挥发,而Vc不易稳定保存,使测定结果易出现偏差,且这种方法不适合微量分析;国标GB/T6195-1986是采用2,6一二氯靛酚滴定法。利用样品溶液由蓝色转变为粉红色来辨别其滴定终点的到达。但是多数水果、蔬菜样品其提取液都具有一定的色泽而导致滴定终点不明显,使测定准确度降低。另外还有荧光光谱分析法 J、紫外一可见分光度法、色谱法、电化学法等,这些方法都存在着一定的局限性,如操作过程复杂,所用试剂不稳定,速度慢、背景¨干扰大。近年来,建立的测定Vc的其他方法还有催化动力学和光度法相结合的方法,及VC传感器测定方法,固定pH滴定法等。 该论文将对蔬菜、水果常用的维生素C含量的检测方法进行综述、比较。 图1 维生素C的结构式1原子吸收分光光度法利用原子吸收分光光度法问接测定维生素C的含量,是利用维生素C可以与一些金属离子发生氧化还原反应,通过测定反应掉的金属离子的量,进而间接计算出维生素c的含量。1.1以银离子作为氧化剂的间接原子吸收分光光度法以银离子作为氧化剂的间接原子吸收分光光度法,是利用维生素C分子中的有二烯醇基具强还原性,可被硝酸银氧化为去氢维生素C,同时产生黑色银沉淀(反应式如图2)。 图2维生素C与银离子反应的反应式 沉淀经离心分离后,将分离得到的沉淀用硝酸溶解后,再利用原子吸收分光光度计测定银离子的含量,从而接测得维生素C含量,具体测定方法如下: 配制一系列浓度的维生素C标准溶液,依次吸取一定量的维生素C标准溶液置于10mL离心管中,分别加入2mL(1mg/mL) Ag+标准溶液,然后加水使总体积为4mL,摇匀,在室温下避光放置35min离心分离弃去上清液,用2mL超纯水洗涤沉淀3次,然后用l:1的浓硝酸3mL溶解沉淀,移入50mL的容量瓶中,加水稀释至刻度。喷入空气-乙炔火焰分别测定其吸光度,以维生素C标准溶液的浓度为横坐标,以测得的吸光度值为纵坐标绘制标准曲线。最后将处理过的待测样按上述方法测定其维生素C含量。 上述方法巾维生素C标准溶液及样品的配制都是利用2%的柠檬酸作为溶剂,并在棕色瓶中保存,原因是维生素C在溶液中不稳定,遇氧气、光、热、碱性物质易受破坏,而在适当的酸性条件下比较稳定,用2%的柠檬酸溶液来配制维生素C标准溶液,减缓了维生素C被氧化的速度,同时消除了一定外界因素的十扰,使得测定结果比较稳定。1.2 以铜离子作为氧化剂的间接原子吸收分光光度法文献中报道了以铜离子作为氧化剂的间接原子吸收分光光度法。该方法也是利用维生素C在酸性介质中维生素C可将Cu2+定量的还原为Cu+,然后Cu+与SCN-反应生成CuSCN沉淀,在高速离心机下有效地分离出CuSCN沉淀,洗涤后再经浓硝酸溶解,用原子吸收法测定沉淀中的含铜量,即可推知样品中维生素C的含量。具体测定方法如下: 分别吸取1mL配制的含一定量维生素C的标准溶液(随配随用)(分别含维生素C 50、100、200、300、400、500 µg)和样品提取液,依次放置于已编号的15mL离心管中,各加入1mLCuSO4饱和溶液、1mL浓度为2%硫氰酸铵溶液、0.5mL盐酸-醋酸钠缓冲液和0.5 mL饱和NaCl溶液充分混和,稍后离心分离,弃去上层清液,小心地用少量水洗涤沉淀2~3次(注意每次用水不能超过1 mL),加入0.5mL硝酸溶解后,转移至lO0mL的容量瓶中加水定容至刻度线,摇匀。分别用原子吸收分光光度计测定其含铜量,由所得的维生素C含量的标准曲线,可以得到相应样品的试验结果。 该方法所得的试验结果相对标准偏差RSD在5%以内加标回收率在96.56%~100.67%,其精密度和准确度均达到痕量分析要求。此方法的线性相关系数R=O 9989,表明相关性很好。2紫外可见分光光度法利用紫外分光光度法测定维生素C的含量是基于维生素c在紫外光区有特征吸收,但是因为维生素C结构中具有不饱和键,具有还原性,不易稳定存在,直接测定误差较大。所以在利用紫外分光光度法测定时,维生素标准溶液和待测样的配制条件非常重要。曾国富,黄玉英研究发现,维生索C在CTAB-C5H11OH-H2O微乳液体系中非常稳定,它存在于微乳液滴膜的内侧,与渗透进入液滴膜外侧的溶液氧接触的机会极少,该体系能极大地提高维生素C的稳定性。 郑京平等利用维生素C具有对紫外产生吸收和对碱稳定的特性,建立了紫外分光光度快速测定水果、蔬菜维生素c的新方法。根据维生素C具有对紫外产生吸收和对碱不稳定的特性,于波长243nm处测定样品溶液与碱处理样品两者吸光度之差,通过查校准曲线,即可计算样品巾维生素C的含量。此法操作简单、快速准确、重现性好,结果令人满意。特别适合含深色样品的测定。实验结果表明该方法简单易行,结果准确、灵敏度高,检出限低,可快速测定水果、蔬菜中维生素C,值得推广应用。 张立科等介绍了在0~450µg/mL线性范围内,以cu2+作催化剂,以溶解氧将还原型维生素C氧化为在246.0 nm处无吸收的氧化型Vc,实现了样品各紫外干扰成分的本底校正,建立了种测定果蔬Vc的新方法。该方法中维生素C破坏条件的选择尤为重要,确定条件为:Cu用量为30 g,反应温度为70℃,反应时间为20min,加热条件下,反应速度快,无需加掩蔽剂。方法简便、快速、准确,测定了香蕉、西红柿等果蔬中的VC含量,结果令人满意。经多次实验得出该方法RsD在0.32%~0.89%之间,实际测定了香蕉、西红柿等样品中的VC的含量,检出限为0.2791g/mL,加标回收率在97.16%~100.18%之间 。3高效液相色谱法前面介绍的方法由于在使用中有一定的限制,操作复杂、前处理较麻烦。因此在使用中有较大的局限性,目的已逐渐被高效液相色谱法所取代。HPLC法具有检测速度快、操作简单、实验结果可靠等特点。 王艳颖,姜国斌等采用HYPERSIL-C8fz谱柱、浓度0.1%的草酸作流动相的高效液相色谱法,分析了草莓中的维生素c含量,取得了理想的效果。HPLC检测条件如下: 流动相0.1%草酸溶液,流速1.0 mL/min;检测波长254 nm,进样量5µL,柱箱温度3O℃。该方法分析中受样品中其他杂质的影响较小。测定草莓中维生素C的含量,回收率为97.4%~102.1%,说明该方法具有所需试剂少、稳定、操作简便等特点。精密度实验的相对标准偏差小于3%,说明该方法重复性和再现性都是比较高的。 陈昌云等采用0.05 mol/L磷酸二氢钾缓冲液:甲醇=80:20(v/v)作流动相。流速为1.0 mL/min,二极管阵列检测器,检测波长为254 nm。测定蜜柚中维生素C含量在质量浓度为20~100mg/L范围内有良好的线性关系,方法回收率为92.4%~107.5%,相对标准偏差小于2%。4 结语测定维生素C含量方法很多,各种方法各有优缺点,因为维生素C自身的不稳定,导致了很多方法测定结果误差较大,所以对维生素C稳定存在条件的探索非常重要。高效液相色谱法因为测定较准确、灵敏度高、选择性好,有较好的发展前景,是目前发展较快的一种方法。
如果对大家有所帮助就好[B]维生素C含量的测定[/B]维生素是是我们经常听到的一个词语,我们每天都要通过食物摄入各种各样的维生素,维生素同我们的健康是密切相关的。维生素C(又称抗坏血酸)普遍存在于水果和蔬菜中,也是一种对人类而言至关重要的物质:人体缺乏维生素C将导致坏血病,维生素C还能防止传染性疾病,甚至癌症。所以,食品饮料、医药、医疗等行业都要测定食品、饮料、药品以及血液中的维生素C的含量。 测定维生素C有多种方法,包括采用I2或二氯靛酚(DPI)进行氧化还原滴定。一般来说,滴定法是一种快速、简便、准确的技术,它通过滴定剂和被滴定物质的等当量反应,精确测定被测物质的含量。DPI对于维生素C具有良好的选择性,是一种理想的氧化剂。 传统的滴定法是手工滴定,根据指示剂颜色的变化确定终点,通过测量滴定剂的消耗量,计算被测物质的含量。手工滴定有很多不足:手工控制误差较大,计算复杂,针对不同的反应需要特殊指示剂。梅特勒-托利多的自动电位滴定仪解决了这一问题,通过测量滴定反应中电位的变化确定终点,全自动操作、计算,测量快速,结果准确。梅特勒-托利多的滴定仪配有记忆卡软件包,存储有成熟滴定方法,可方便快速解决实际应用问题,并且稍作改动就能作为新的测定的实验方法。以下是某饮料企业使用梅特勒-托利多TX电位滴定仪测定饮料中维生素含量的实例样品来源样品量gVc含量(mg/100g)aVc含量(mg/100g)bVc含量(mg/100g)cVc含量(mg/100g)dRSD%猕猴桃新鲜切片5103.79 104.25104.81 103.960.979橙汁1瓶装537.04 37.1637.21 37.090.507橙汁2瓶装537.19 37.0637.03 37.120.501橙汁3瓶装537.14 37.0437.09 37.200.447鳄梨汁1瓶装541.46 41.5241.65 41.560.477鳄梨汁2瓶装541.58 41.5441.64 41.460.434柠檬汁1鲜榨553.00 53.1853.12 53.090.339柠檬汁2鲜榨553.01 53.1353.06 53.180.321柠檬汁3鲜榨553.13 53.1752.96 53.080.396葡萄汁1瓶装1053.04 53.0553.04 53.050.022葡萄汁2瓶装1053.03 53.0653.05 53.050.057葡萄汁3瓶装1053.05 53.0653.06 53.040.044[~192934~]
液相色谱-串联质谱法检测食品中维生素D含量 如何用液相色谱-串联质谱法(LC-MS/MS)检测食品中的维生素D含量。这项技术可是现代食品分析中的佼佼者,能够帮助我们精准地掌握食品中的营养信息。 样品前处理 提取:首先,我们需要从食品样品中提取维生素D。这一步很关键,因为提取效率直接影响最终的检测结果。通常,我们会使用有机溶剂,比如乙腈或甲醇,来提取维生素D。 净化:提取后的样品往往含有很多杂质,这些杂质会影响检测结果。因此,我们需要对提取液进行净化处理。常用的净化方法有固相萃取(SPE)和液液萃取(LLE)。 浓缩:净化后的样品溶液需要进行浓缩,以提高维生素D的浓度。常用的浓缩方法有氮气吹干和旋转蒸发。 仪器操作 流动相选择:选择合适的流动相对分离效果至关重要。通常,我们会使用水和有机溶剂(如甲醇或乙腈)的混合物作为流动相,并根据需要添加少量酸或缓冲液。 色谱柱选择:选择适合的色谱柱也很关键。C18反相色谱柱是常用的选择,因为它对维生素D有很好的保留效果。 质谱条件:设置合适的质谱条件,包括离子源温度、喷雾电压、碰撞能量等。这些参数的优化可以大大提高检测灵敏度和特异性。 故障排除 峰形不好:如果发现峰形不好,可能是由于流动相比例不合适或色谱柱污染。尝试调整流动相比例或清洗色谱柱。 灵敏度低:如果灵敏度不够,可能是由于样品提取效率低或仪器参数设置不当。检查提取方法并优化仪器参数。 杂峰干扰:如果出现杂峰干扰,可能是由于样品净化不彻底或流动相选择不当。尝试改进净化方法或更换流动相。 仪器故障:遇到仪器故障时,首先要保持冷静,然后根据仪器的报错信息查找原因。必要时,可以联系仪器厂家进行维修。 总之,液相色谱-串联质谱法检测食品中维生素D含量是一项复杂但非常重要的技术。通过掌握这些操作要点和故障排除方法,我们可以更加准确、高效地完成检测任务。
注射用水溶性维生素含量检测各位高人,请教一下:我所检测的样品含:烟酰胺,泛酸钠,VB1,VB2,VC,VB6,VB12,叶酸和生物素及对羟基苯甲酸甲酯用的液相,由于没有荧光检测器,核黄素磷酸钠就用其中核黄素就用了烟酰胺的方法,用紫外检测器检测,烟酰胺,泛酸钠,VB1,VB2,VC,VB6为一组,条件:30度,214nm;叶酸、生物素一组,条件:30度,200nm,对羟基苯甲酸甲酯和叶酸组条件一样,只是流动相乙腈比例较大;VB12 用的是梯度洗脱,40度,360nm。其中核黄素磷酸钠和生物素不好测,请问各位有没有更好的方法呢?补充:我用紫外扫描,发现核黄素磷酸钠在200nm出无吸收,但是VB1也无吸收,请问吸收度于出峰时间有事么关系呢,是否同一波长下吸收度大的出峰早,峰面积大呢?谢谢!
请问大家,哪位高人有测维生素E含量简单有效的方法,最好是HPLC方法,国标的方法太麻烦了,药典是气相自己做不了!
[em0801]红外光谱测定地产小麦维生素E含量 这个我没弄懂它的原理,还不确定检测它的方法,所以在此发出求助的帖子,希望高手指导,小妹谢拉!!
按照国产方法测预混料里维生素b12 的含量样品峰分不开,应该怎么办
请教:液相色谱如何分析维生素C的含量,是饲料中的维生素C,样品如何处理。操作步骤。谢谢各位老师指点迷津。
1.利用C18柱反相高效液相测定维生素D3的含量时流动相能否加入缓冲液?一般加入的缓冲液有哪些?以及缓冲液的ph值和所占流动相的比例是多少?2.利用C18柱反相高效液相测定维生素D3的含量时,它的含量如何计算?计算公式是什么?如何推导?与中国药典2010版有什么不一样?影响因子在反相高效液相中如何测定以及计算?3.测定维生素D3含量时,对照品的单位是mg,而样品中含有的维生素D3的单位是国际单位(IU),在配制溶液时,单位是否要换成一致,对后面的含量计算有什么影响?一般维生素D3在上反相液相是进样所配制的浓度范围是多少?
哪位大侠用紫外分光检测过新鲜的蓝莓等水果中的维生素C的含量吗?我查到的文献中用氢氧化钠破坏维生素C测得到吸光值与未破坏时测得的差值,即为还原型维C的含量。但是我做的时候在243nm测吸光值,用氢氧化钠破坏维生素C测得到吸光值比未破坏的还要大,是什么原因呢?
二阶导数分光光度法测定维生素E霜的含量 作者:张洪 马俊玲 周健 单位:湖北医科大学附属第一医院药学部,湖北武汉 430060 关键词:二阶导数分光光度法;维生素E;药物分析 广东药学院学报000210 摘 要 用二阶导数分光光度法测定维生素E霜中维生素E的含量,可消除基质对测定的干扰,方便简便、快速且准确。回收率为98.24%(RSD=0.61%)。 中图号 R927.2 文献标识码:A 文章编号:1006-8783(2000)02-0112-03 维生素E霜主要成分为维生素E(Ⅰ),现行测Ⅰ含量标准[1]采用[url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]法,由于基质的干扰,无法按标准方法进行含量测定。我们根据Ⅰ的紫外吸收特点,用二阶导数分光光度法消除基质的干扰,不经提取分离,直接测定Ⅰ的含量,方法简便、快速、准确可靠。适合医院对该制剂质量控制的要求。
维生素C的含量测定的方法,尽量详细一些,最好有滴定、光谱、色谱等。。。
我们现在用国标中检VB2的方法检B族维生素(主要是VB1,VB2,VB6),检原料及维生素含量超过1%的饲料都能检的很准,但是检测含量只有几十上百个ppm的预混饲料时就很难检准,主要是杂质太多了,图谱的基线都不是平的,峰很难判断。大家有没有比较好的方法啊。 我最近想通过离子交换的方法去杂,也做了些试验,比如用PCX小柱做萃取净化,只有VB6的效果比较好,VB1和VB2都有比较高的损失,有没有人做过这方面的尝试啊?有的话分享一下经验呗!
哪位大侠检测过核桃粉中Vc的含量?不知道用《婴幼儿食品和乳品中维生素C的测定》这个方法行不行?
我用GB的方法测定乳品中的维生素K1的含量,可是在前处量后用氮气就吹不干,而且分析时不出现任何峰.不知是什么原因?难道与脂酶有关吗?请大家赐教?
维生素C含量高的蔬菜:[img]https://ng1.17img.cn/bbsfiles/images/2019/08/201908051809445366_604_1241901_3.png[/img]
安利纽崔莱维生素C爆出含量不达标,可能美国人不会检测,迪马给他提供Vc的检测方案http://simg.instrument.com.cn/bbs/images/brow/em09503.gifhttp://ng1.17img.cn/bbsfiles/images/2012/07/201207271104_380215_2370618_3.jpghttp://ng1.17img.cn/bbsfiles/images/2012/07/201207271104_380216_2370618_3.jpghttp://ng1.17img.cn/bbsfiles/images/2012/07/201207271105_380217_2370618_3.jpg
作者:http://d.g.wanfangdata.com.cn/Images/head_pic.gif吴琳 作者单位:湖南省衡阳市药品检验所,421001 摘要: 目的:建立反相高效液相色谱法测定维生素B1片含量的方法.方法 :采用DLamonsLlTM(钻石)色谱柱C18(250×4.6mm,5um);流动相为0.05mol/L辛烷磺酸钠的1%冰醋酸溶液-甲醇-乙腈(50:30:20);流速:1.0ml·min-1;检测波长:254nm.结果 :维生素B1在50~350ug·ml-1范围内线性关系良好,平均回收率为99.6%,RSD=0.9%(n=7).结论 :该方法简便可行、结果准确可靠,可作为维生素B1片的质量控制.
[em0813][em0812]有没有人用国标的方法做过维生素D含量测定呀,最近我在用这个方法做,遇到点问题。是不是在做前处理的时候要完全在暗处操作,还有在计算的时候要不要把图谱上面所以的峰都算进去?谢谢各位了。
有人用高效液相做过乳品中维生素A、D、E的含量检测吗?一起来探讨一下吧!
制备的盐酸硫胺(即维生素b1)测折干含量和折水含量都到120%左右,为什么?
作者:胡少卿; (广东省人民医院药学部;)摘要:目的:建立高效液相色谱法测定复合鱼肝油制剂中维生素A含量的方法。方法:采用高效液相色谱法,选择迪马(Diamonsil)C18(4.6 mm×250 mm,5μm)色谱柱,流动相为:甲醇-水(95∶5),流速1.0 ml/min,检测波长为326 nm,柱温为35℃。结果:维生素A进样量在2.907 0~34.883 7 IU/ml范围内呈良好的线性关系(r=0.999 6),平均回收率为98.84%。三个批次的复方鱼肝油制剂的含量分别为62.082、62.163、62.558 mg/瓶。结论:以高效液相色谱法检测复合鱼肝油制剂中维生素A的含量,结果准确、重现性好,精密度高,可作为该制剂的质量控制方法。谱图:无
[font=&]检测100万IU/g维生素A醋酸酯原液,用5009.82检测,检测到维生素A是一半左右,是大部分维生素A醋酸酯在检测过程中不能转换成维生素A吗[/font]
请教各位师兄师姐,岛津的液相可以分析维生素C的含量吗?用什么流动相。柱子。是否需要柱温箱?
中国药典规定,维生素E的含量测定用[url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]法做,其实,液相是非常方便的。但是标准定了,只能执行。在实验过程中,发现柱子的选择很重要,用HP-5的柱子,200度20分,5度每分到240度,再保持25分,出峰需要1小时左右,分离度勉强达到1.6(与内标正三十二烷)。有没有做过实验的,告诉我什么柱子更好?HP-1的怎样?
各位老师, 您好,我们单位生产功能性饮料, 需要检测维生素B2,当然里面还有其他维生素B1,B3,B5,B6等。我们使用过的是GB5009.85,仪器是岛津的2030PLUS,荧光检测器。现在我们在检测过程中,发现检测值和实际添加值相差很大,比如实际添加1.0,检测值才0.2。不知道哪里出问题了样品前处理大致流程是:称样液5ml,加60ml0.1盐酸水解,121℃高压灭菌30Min。取出冷却后调PH至6.0-6.5,加2ml混合酶溶液,培养过夜,第二天拿出定容100ml,离心去上清液过滤上机。检测添加剂稀释后的含量值和标准相符。产品不水解酶解直接稀释检测,结果0.44疑惑:水解和酶解的主要作用是?
我们公司正在开展乳粉中维生素A、E的检验工作,但是按照最新的国标方法出现以下几个问题,希望各位指点一下1、看到别的方法上无水乙醇需去醛处理,我们用的色谱纯的乙醇还用处理吗?2、其他方法上有用乙醚萃取的,而我们用的是石油醚是否对结果有影响?3、其他方法上有用乙醚萃取的,乙醚需检测是否含过氧化物,石油醚是否用检测?4、最重要的是我们用国标液相法做完后,回收率一直在40%多,做已知含量的样品,检出值也是原值的40%左右。我们完全按国标方法做的,不知什么原因,望做过的大侠们指点!
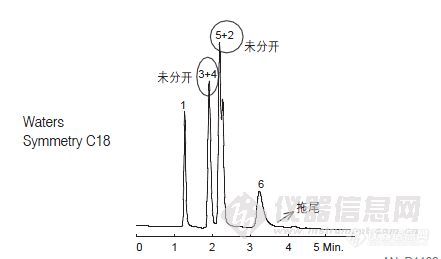
我们现在做维生素的检测,遇到以下几个问题,希望高手门能给于解答。1.标液出峰的问题:我们现在配的是混标(VA、VD两种、VE三种)维生素A是完全没有问题,但是VD,VE这5个峰有点分离效果不太好,浓度低时这5个峰是完全可以分开的,但是一旦样品中这5种物质一高就会有部份重叠了。2.加标准回收的问题,现在只是用空白做加标皂化后回收率都只有20%,请问一下是哪里出了问题?3.做这些维生素的有没有什么地方要特别的注意的







 我要推广仪器
我要推广仪器
 下载APP
下载APP