请教!实验室中[url=https://insevent.instrument.com.cn/t/Mp][color=#3333ff]气相[/color][/url]检测纯甲醇、纯乙醇的纯度时,经常会出现样品平行样里的某一针峰面积响应突然增大(约正常的面积的一倍)。目前衬管、隔垫、进样针及色谱柱均换了新的依然出现问题,未找到明显的规律性。论坛上的大佬们给指点一下!
做标准时峰面积和峰型都很好,但进入被检测样品时峰面积又矮又小。求解
这两天经常做同一个样品,对照品叫栀子苷,昨天用VWD检测器分析,对照品的峰面积有600多点,样品含量很正常,今天用DAD检测器分析,峰面积只有300多点了,样品含量也很正常!对照品溶液很稳定,是不同的检测器对有些物质的响应值不一样啊?
我用的是GC-14C,FPD检测器,DB-1毛细管柱,柱子已经用1年多了,检测器也用好多年了。最近突然样品峰面积变为原来的一半,峰型没什么变化,溶剂峰面积没有变,但是分离度不好了。而且最低只能测到0.1mg/L了。因为是学生刚开始做,[url=https://insevent.instrument.com.cn/t/Mp]气相[/url]几乎不懂,请大家帮帮忙吧,帮我分析一下怎么回事。
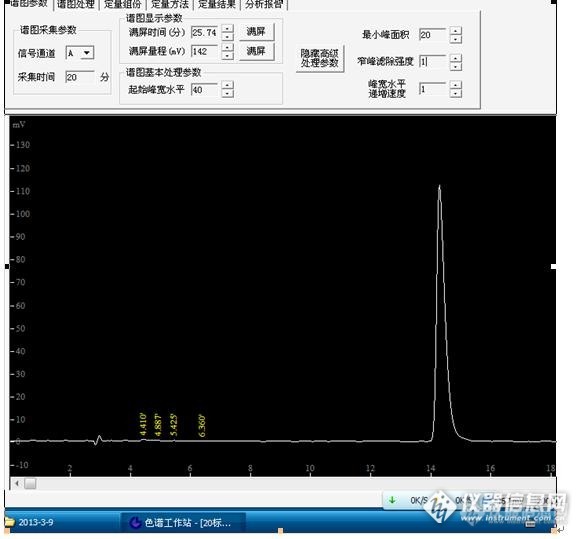
各位高人:本人用高效液相色谱分析盐酸小檗碱时,标准 品走样时在4-5min有一个小峰,检测到了,但是13-15min的大峰反而检测不到,没办法计算峰面积,这是怎么回事?有哪位高人给指点一下,该如何计算其峰面积?http://ng1.17img.cn/bbsfiles/images/2013/03/201303141458_430196_2583865_3.jpg
各位老师,想问一下cma对认可第三方检测,对实验室的面积有什么要求没有
请问下,杭州、上海有没提供测粉末粒度分布和比表面积的第三方检测机构或单位?要求盖公章的。
使用安捷伦[url=https://insevent.instrument.com.cn/t/Mp][color=#3333ff]气相[/color][/url]顶空进样检测样品中的乙酸残留,检查结果图谱乙酸衍生物峰面积比正常值小了三分之一,其他峰面积跟正常值一样,哪位老师能给解答一下
使用安捷伦[url=https://insevent.instrument.com.cn/t/Mp][color=#3333ff]气相[/color][/url]顶空进样检测产品中乙酸,检查结果图谱乙酸衍生物峰面积比正常值小了三分之一,其他峰面积跟正常值一样,哪位老师能给解答一下,什么原因导致的。
我用的是上海天美7890[color=#333333][font=宋体]Ⅱ 检测器用FPD 分析沼气中的硫化氢 百分含量 0.1-0.5% 开机后半小时之内分析没有问题,时间长了峰面积明显变小,等降温后在打开从新升温点火又恢复正常,时间一长又变小,不知道什么原因。氢气氧气流量比大概4:1,检测器温度200 色谱柱是毛细柱 柱温 100 进样口 180 是双火焰检测器,还有就是如何定量。是浓度与面积平方成线性还是浓度的指数与面积的指数成线性?[/font][/color]
萘甲醇1×10-7荧光检测器峰面积大约是多少,保留时间是多少,有没有做过校准的小伙伴儿
在做蔬菜中氨基甲酸酯类农药的检测,用的是岛津LC-20A,检测器用的是RF-20A荧光检测器(氙灯)。因为随时都可能有样品要做,所以仪器基本是不关的。最近十个农药峰的峰高已经从开始的平均15.0mv降至平均1.0mv,峰面积也降的很厉害(峰型正常)。看了一下检测器的使用时间也超过了限定的2000h,所以就更换了检测灯。原以为更换了检测灯以后峰高和峰面积都会回到初始值,但这几天走样下来发现还是老样子,峰高也没升多少,维持在平均1.5mv左右(峰型正常)。 哪位朋友能解答一下到底是什么原因呢,导致峰高和峰面积值下降的原因是不是由检测灯的使用时间增长引起的?如果是的话那为什么换了灯以后峰高和峰面积值没有回到初始值?还有跟色谱柱没有换是否也有关系呢?
同一种物质,前后检测两次为什么得到的峰面积会不一样??有时候甚至有10000多的差异……
FPD检测器峰面积与样品的浓度成正比还是与样品浓度的平方成正比!谢谢![em06]
便捷式非甲烷总烃检测器,内面的色谱柱加热方式是什么请问,用加热丝吗,毕竟是用锂电池供电的,那功率且不是只能选小功率,加热那不是要很久?[img=,234,269]https://ng1.17img.cn/bbsfiles/images/2019/07/201907100123176437_5779_3947244_3.png!w234x269.jpg[/img]
更换载气后,基线漂移(最高达300pA)。请问各位,GC如何设置 可提高检测物种响应值(如峰高/峰面积)?理论上,是否检测物种响应值增大,即便基线漂移,对检测物种峰面积积分的影响也可一定程度上缓解?【例如检测物质峰高基本2000-3000 pA,即便基线漂移最高至300pA,其影响程度也低检测物质峰高基本500-1000 pA(基线漂移最高至300pA)】
各位前辈,初学GC,遇到一次仪器故障,外标法测组分的过程中,标准物进针,发现面积与以前做的增加了三倍,进针两次都是这样,最后发现是进样口漏气。这个过程中在进样前发现压力变小了,只是简单地调大了,气压螺丝拧了不怎么变化,,因为不熟悉仪器,只是认为气压不好控制。在分析过程中,因为每次的分析时间短,压力变化不明显,保留时间变化也不大,只是峰面积增加了几倍。现在本人的疑问是GC检测中,压力和峰面积存在什么样的关系,所用的检测器的FID。谢谢!
TCD是浓度型检测器,是不是说同一浓度不同体积进样,色谱峰高或者峰面积不变呢,求解答
明胶空心胶囊检测环氧乙烷残留供试品峰面积大于对照品峰面积,不知道怎么回事?已经做过定性定量分析,确定就是环氧乙烷的峰,但是测样品胶囊时,出来的峰比对照品峰面积要大,我拿的是未经过环氧乙烷灭菌的胶囊,应该说是检测不出环氧乙烷的。生产环境里也没有环氧乙烷啊,不知是哪出了问题?求高人指点!胶囊检测标准在中国药典2010年版二部第1205页,谁能帮我研究研究。对照品溶液的浓度为0.2ug/ml,对吧?还有一个问题,做顶空进样的空瓶也能检出峰来,我已经问过很多专业人士了,都找不出原因。
RT.例如:A和B混合,用火焰检测器做出的峰面积比是40:60.那是不是意味着A的含量是40%。如果不是那么火焰检测器检测的依据是什么?求助!!!!!!!
[size=2]六六六滴滴涕检测 ECD检测器 峰面积明显变小!我做六六六滴滴涕的实验时,第一天进标样出峰情况正常,第二天所有条件都没变,进了一针同样的标样,峰高明显变小了,而且是馒头峰,仪器也是刚刚调试过的,检测器应该没有问题。请问有可能是什么原因呢?仪器具体条件是:山东鲁南SP6890型[url=https://insevent.instrument.com.cn/t/Mp]气相色谱仪[/url],农残二号柱子,柱温185℃,汽化210℃,检测230℃,恒温运行,尾吹0.06,柱头压0.06,分流和灵敏度都是调试过的,基本都不用调。[/size]
我用的检测器型号和厂家不祥。做的是内酯的检测,用的是梯度洗脱,主要溶剂是甲醇和水。在检测过程中发现同一天在不更换流动相的情况下,同一样品连续进样,峰面积随着检测时间的增加,不断增加。经过排除,可以排除泵、溶剂挥发、样品等问题,因为不是常用这个检测器,所以不知道检测的问题出在哪里,请个位大虾帮忙!
导致组分在FID检测器上的峰面积减小的因素有哪些?(1)从一次更换检测器时发现,同一浓度的组分在新的检测器比旧检测器峰面积响应大。(2)喷嘴部分堵塞。欢迎给位拍砖,并加以补充!尾吹的大小是否也是一个因素呢?
请教高手,我用FPD检测有机磷乐果,溶剂峰峰高异常地高,乐果的峰面积却很小很小,用的载气是高纯氮气,氢气压力0.1,空气压力0.2,柱压0.15
1.[b]主题:【分享】2024年1月份纺织品检测版面积分发放[/b]https://bbs.instrument.com.cn/topic/8316983 2.[b]主题:【分享】2024年2月份纺织品检测版面积分发放[/b]https://bbs.instrument.com.cn/topic/8323743 3.[b]主题:【分享】【分享】2024年3月份纺织品检测版面积分发放[/b]https://bbs.instrument.com.cn/topic/8334299 4.[b]主题:【分享】2024年4月份纺织品检测版面积分发放https://bbs.instrument.com.cn/topic/8346946[/b] 5.[b]主题:【原创】2024年5月份纺织品检测版面积分发放[/b]https://bbs.instrument.com.cn/topic/8357750 6.[b]主题:【原创】2024年6月份纺织品检测版面积分发放[/b]https://bbs.instrument.com.cn/topic/8371517 7.[font=&][size=16px][color=#333333]主题:【原创】2024年7月份纺织品检测版面积分发放[/color][/size][/font]https://bbs.instrument.com.cn/topic/8387501 8.[b]主题:【分享】【原创】2024年8月份纺织品检测版面积分发放https://bbs.instrument.com.cn/topic/8403613[/b]
检测同一个样品,两次进样吸收峰面积差别很大(手动进样,5ul定量环)原因是什么?请高手指教。共两个峰,一次是17500000/38590000,第二次是10986000/2434000.
[url=https://insevent.instrument.com.cn/t/Mp]气相色谱仪[/url]检测器是FID检测器,进样0.5ml,为什么只是高浓度进样才有峰面积,低浓度进样没有峰面积?真心求解答,谢谢各位
在气相分析中常用到面积归一化法定量,但现在有一个困惑:遇到未检出的组分,是直接报未检出还是报小于检出限?比如,有一混合物有A、B、C、D四种组分,用面积归一化法定量,即A+B+C+D=100%。若A组分检测不出来,就是B+C+D=100%,这样的话,A在检测报告中,该发怎样的结果呢?报未检出的话,不符合要求;报小于检出限,感觉和面积归一化法有冲突。到底该如何呢?
[align=center]液相面积归一法如何确定波长[/align]摘要:一般现在液相检测通常有面积归一法、校正面积归一法、外标法、内标法等几种方法,而检测纯物质,一般只采用面积归一法,而在确定杂质并提取到杂质对照品或提取到主成分对照品时,也可采用加校正因子或不加校正因子的主成分自身对照法进行检测。但是在实际检测过程中,有时候会出现波长确定比较难的情况,本文就如何在使用面积归一法时确定波长并且在主成分波长和杂质波长不同时如何进行选择提出可行性方案。 在使用液相进行检测一种物质的时候,如果没有标准方法、文献可供参考,我们需要通过以下几种方法进行判定波长,第一种是使用DAD二极管阵列检测器,可以很方便的确定被测组分,包括主成分和相关物质的最大吸收波长或者特征波长;第二种是通过紫外可见分光光度计进行波长的扫描,也能得到被测主成分的最大吸收波长或者特征波长,但是无法判定相关杂质的最大吸收波长,而且液相并不一定使用的是最大吸收波长,还可能从分离度等方面进行考虑。第三种是液相紫外检测器有波长扫描功能的,通过扫描波长进行确定目标组分的吸收波长,缺点仍然是只能一个波长测试一遍,才能进行分离度等其他参数的考察。 一般面积归一法测试进行选择波长的过程中,优先选择被测组分的最大吸收波长,其次是特征波长,并且要确定流动相在该波长的吸收不对被测成分产生干扰,选择波长最好比溶剂波长高20nm左右,这样干扰就比较小。但是单从面积归一化法这一个检测方法进行考虑,它是无法判断样品中相应成分的准确含量的。如果合成的相关化合物结构比较类似,都有紫外吸收,其紫外吸收曲线比较一致,有一定的参考价值。但一般情况下,其可靠性是很差的。但是,对于纯物质的判定,往往不能通过外标和内标法进行测定,而是采用面积归一法结合其他方法进行测定。如果出现纯物质的检测,主成分和相关杂质的在一个波长上响应不同,例如:该样品主成分在213和261nm处有两个吸收峰,213nm处为最大吸收峰。213nm测得的纯度较261nm低2%左右,杂质的出峰情况不同。紫外谱图如下:红色为213nm谱图,黑色为261nm谱图[img=,500,215]https://ng1.17img.cn/bbsfiles/images/2019/10/201910240958105464_2581_3295053_3.png!w500x215.jpg[/img][img=,500,312]https://ng1.17img.cn/bbsfiles/images/2019/10/201910240958108954_5224_3295053_3.png!w500x312.jpg[/img] 我们首先可以对主成分及主要杂质进行质谱分析,确定其化学式及其化学性质,如果有该杂质的对照品或者该杂质能够被提取出来,我们可以做杂质与主成分的校正因子,那么在某个波长下,该杂质的相对于主成分的校正因子满足0.8-1.2的范围内,我们就可以确认这个波长是适合的。 如果遇到比较难提取的杂质,但是主成分有相应的对照品,也可以在不同波长下进行主成分对照法进行,配制对照溶液并调节仪器灵敏度后,取供试品溶液和对照溶液适量,分别进样,测量供试品溶液色谱图上各杂质的峰面积,并与对照溶液主成分的峰面积比较,计算杂质含量。通过不同波长下面积归一法与主成分对照法测试的杂质含量的比值进行确定最佳检测波长。如果主成分也没有相应的对照品,这时可以采用衍生化法,不过这种方法仍需了解主成分及相关杂质的化学性质等,然后采用合适的衍生剂,将主要杂质和主成分用衍生的手段把吸收波长调整至非常接近,然后采用相同波长进行检测。这种需要的专业知识较高,而且衍生化法在实际应用中也存在衍生剂干扰、衍生不完全等情况。 还有一种方法是在通过NMR的氢谱、碳谱等相关的谱图先基本确定样品很纯的情况下,通过于其他检测器或者检测方法进行比对,比如我们可以通过卡尔费休法检测水分、顶空色谱检测残留溶剂、灼烧测定灰分、[url=https://insevent.instrument.com.cn/t/3p][color=#3333ff]离子色谱[/color][/url]测定一些小分子有机酸,然后结合紫外分光光度计、化学滴定等方法进行主成分的检测,并且与其在液相色谱上的各相关波长结果进行比对,即可得出仪器检测的最佳波长。综上所述,面积归一法并不能只看一个波长的结果,为了得到可靠的结果,我们需要综合判定其在不同波长下的结果进行比对,并根据主成分和杂质的波长响应,得到最佳检测波长,有条件的情况下,还需要通过其他检测器和检测方法进行比较。
用液相紫外检测器测试标准品(产品)纯度,面积归一方法,取样量多少合适?在应用中样品多一点,色谱柱分离不好,峰型变形,结果偏低,稀释后峰型完美了,结果也高了,平行测定结果偏差较大,哪位在生产岗位应用液相做品控,给提供个最佳取样量?







 我要推广仪器
我要推广仪器
 下载APP
下载APP