水分散粒剂中分散性是很重要的一项指标,由于检测设备不齐全,我们一般把悬浮率高的认定为分散性也就没问题,大家是怎么看呢?农药分析上写的,悬浮率是颗粒自然分散后的结果,而分散性是给一定的外力看分散性如何?既然这样,那么如果不给外力都能达到很高的标准,那么加外力后分散效果肯定是好啦?可以这么理解吗?
采用液相色谱一二极管光谱检测器一串联四级杆质谱(LC—PDA—MS/MS)同时快速测定纺织品中的22种有害分散染料。研究设计的氯苯蒸气回流提取法的提取效率是超声波辅助甲醇提取方法的2.3~11倍,采用的1.8Ixm细粒径液相色谱柱比传统的5m色谱柱的分析时间缩短了近三分之二,而检测灵敏度至少提高了4倍。通过电喷雾串联质谱鉴别,确认DIN54231标准中的分散蓝35(b)物质为分散蓝26染料。22种分散染料的测定低限在0.8~5mg/kg之间,回收率均在86.4%~98.7%之间,相对标准偏差值小于10%。在面料、服装衬里布以及缝纫线和拉链边布等一些辅料中都检测出有害分散染料,检出率较高的是分散黄23和分散橙37/76。分散染料是在水溶液中呈分散状态的染料总称,主要用于聚酯、聚酰胺和醋酯等化学纤维及其混纺制品的染色。分散染料对人体皮肤的致敏性一直备受关注,20世纪7O年代报道的“锦纶丝袜过敏”事件就缘于锦纶纤维上的分散染料造成接触性皮炎川。据报道,三分之二的纺织品致敏事件由分散染料造成。目前有20种常用于纺织品染色的分散染料被确认为致敏染料,生态纺织品标签OekorexStandard100和欧盟Eco—label(EU2002/371/EC)都要求纺织品中不得检出这20种染料。由于分散黄23和分散橙149易分解生成具有致癌致敏性的对氨基偶氮苯,Oeko—texStandard100从2006年起要求纺织品中也不得检出这2种染料。分散染料常用的检测方法有薄层色谱法(TLC)、高效液相色谱法(HPLC—DAD)、液相色谱一质谱法(LC—MS)和液相色谱一串联质谱法(LC—MS/MS)。2005年9月,德国标准化协会颁布了标准检测方法DIN54231,规定用超声波辅助甲醇方法提取和液相色谱一二级管陈列检测器一质谱(LC.DAD—MS)方法检测纺织品中的9种分散染料。分散染料为分子质量相对较小的脂溶性染料,性质相近,而且染料中常含有合成中间体、同分异构体和分散剂等杂质;故这22种分散染料的检测要求检测仪器的色谱分离性能较高,检测人员的定性鉴别能力较强。本文用1.8txm的细粒径填料色谱柱,在超高压条件下快速、高效分离22种有害分散染料,根据染料的光谱吸收特征和串联质谱的结构信息进行准确测定,同时,用氯苯蒸汽回流萃取纺织品中分散染料。
分散液-液微萃取是2006年才问世的一种新型微萃取技术。它基于使用微量注射器将微升级萃取剂快速注入样液内,在分散剂-水相内形成萃取剂微珠,很大地扩展了有机萃取剂和水样之间相接触面,大大加快了萃取平衡的速度,使目标化合物迅速萃入萃取剂微珠内,提高了萃取效率和富集倍数。萃取富集操作与后续检测方法有很好的相容性,萃取相可直接进样气相色谱、高效液相色谱、薄层色谱、气相色谱-质谱、分光光度、火焰原子吸收光谱和电热原子吸收光谱,对目标化合物进行测定。它不仅适用于痕量有机物的分离富集,也适用于痕量无机金属离子的分离富集,已成功地应用于各种环境水样(包括高含盐量水样)、多种饮料、生物样品、煤和矿物样品中痕量组分的分离富集,是一种有发展前途的环境友好的分离富集技术。 附件请参考。
目的 建立分散液液微萃取-气相色谱-质谱联用法快速测定饮用水中一氯一碘甲烷、碘乙腈、二溴碘甲烷等7种挥发性碘代消毒副产物。方法 水样加入无水硫酸钠后迅速注入经混合的二氯甲烷及甲醇溶液分散萃取,涡旋震荡并离心后,取二氯甲烷沉积相直接进样,经DB-5MS毛细管气相色谱柱分离,采用选择离子监测模式检测7种碘代消毒副产物。结果 采样二氯甲烷作为萃取剂、甲醇为分散剂提取饮用水中7种挥发性碘代消毒副产物,方法富集因子为69~129,7种物质在DB-5MS色谱柱上分离良好,各物质线性范围为0.3~50μg/L,相关系数均大于0.996,检出限为0.16~1.42μg/L,定量限为0.48~4.26μg/L,低、中、高三水平加标的平均回收率为75.2%~119%,相对标准偏差4.1%~10.5%。结论 建立的方法采用分散液液微萃取提高了水样的提取效率,该方法高效简便、灵敏准确,可用于饮用水中7种挥发性碘代消毒副产物的快速检测。
前段时间我们参加了IIS的检测纺织品中20种分散染料,检出3种,分别是分散3,分散橙1和分散黄3,但是近来那边发来邮件说我们测的分散黄3值偏高,请问你们做出的值大概是多少啊?
我们刚刚接手的,昨天被培训了半天,请问大侠们紫外-可见分光光度计能检测样品的分散度吗?

在农残检测中,首先要面临的问题就是如何把目标物尽可能完全高效地从样品中提取出来,目前提取的常用方法有浸泡(震荡)、超声辅助萃取、均质提取、索氏提取等方法,其中广泛高效应用的应该就是均质提取了,而均质提取最常用到的就是IKA T25分散机,所见过的十几家检测机构都用到它。从我们十年来的使用情况来看,IKA T25分散机确实很好用。分散的目的是将不可溶的固体、液体或气态物质分散到液体中,在农残检测中是让农药目标物均匀分散于有机溶剂中完成农药的提取步骤。T25分散机主要由支架、支臂、固定螺丝、分散机主机、分散刀具组成。分散机带动分散刀具完成均质操作。装配完成如下图一。http://ng1.17img.cn/bbsfiles/images/2012/12/201212200925_414029_1623180_3.jpg分散刀头外壳为定子,内部径向轴承带动的分散刀具为转子。由于转子的高速运转,被分散的介质被自动吸入分散头,然后这些介质呈放射状以较高速度通过转子与定子之间。施加在分散介质上的巨大加速度产生极大的剪切力。另外,定-转子间介质的高速旋转扰动也促使介质达到最佳的分散效果。分散效果很大程度上取决于剪切梯度以及颗粒在剪切区域停留的时间。通常数分钟的分散就可以达到均质所需的效果。长时间的分散仅对于可得到粒度范围内介质的粒径起到明显的改善作用,但过长时间的分散会使介质的温度升高。因此在一定转速下的均质分散并不是持续的时间越长越好。http://ng1.17img.cn/bbsfiles/images/2012/12/201212200926_414030_1623180_3.jpg图二:IKA T25分散机工作原理图IKA T25分散机(基本型)调速轮共有五档分别对应6500转/分、9500转/分、13500转/分、17500转/分、21500转/分。因为分散刀具的高速运转时极容易导致介质液体溅出(在农残检测中就可能造成目标物的损失),所以一般进行分散均质操作时只要使用1-3档就可以满足均质提取的要求。我们主要按以下时间顺序进行农残检测的均质分散操作的:开机1档 30秒2档 60秒3档 30秒2档 60秒1档 30秒关机4档、5档只有在用水洗分散刀具清除样品嵌留时才能用到。为了尽可能保持分散机的优良性能,延长它的使用寿命,以下正确的使用和维护是必不可少的。1、均质器只适用于对处理过程中产生的能量不发生反应且可能产生危险的介质。同时被处理的介质也不能与其它方式产生的能量反应并产生危险,比如光照等。不能用均质器处理易燃、易爆介质,或在易燃易爆的环境中操作仪器。处理易挥发溶剂,要在通风橱中进行。2、使用电源电压必须和铭牌所示的额定电压一致。3、注意经过长期使用,存在磨损,转动部件可能会脱落。仪器震动可能导致固定螺丝的松动,仪器使用前要检查有无损坏或存在安装不牢固的现象,松动的部件要重新牢固安装,拧紧,将仪器牢固固定在支架的竖杆上,操作时要在平稳、防滑、干燥的台面上进行,确保仪器在操作过程中支架台不会移动。确保盛放介质的容器平衡,确保旋转刀具安装到位,固定旋钮安装牢固,并拧紧旋钮可靠固定刀具。确保整个系统良好的稳定性。4、在使用中要防止外部介质、液质渗入仪器内部。5、根据处理介质的种类,操作时要佩戴合适的防护设备。操作时分散转速不要设置太高,以防止介质液体溅出,对身体、衣物及周围物件造成污染或破坏。6、不同仪器的分散器具不要随意混用,以防意外发生。7、不要操作没有旋转刀具的仪器。也不要在干态下操作仪器,没有介质的冷却作用,垫圈和轴承可能会被严重磨损。8、在调试仪器前,确保仪器处于最低转速,然后逐渐提高转速。否则仪器将以设定的速度高速运转,如果由于转速过高导致被分散的介质溅出容器,要降低仪器转速。9、开启仪器前,确保分散刀头浸入待处理介质45mm以下;容器中待处理介质的深度不得少于55mm;分散刀头距容器底部不少于10mm,防止介质液体溅出。10、不是手持型的,不能用于手持操作。固定搅拌容器以防旋转,慎用玻璃容器装待分散介质,玻璃容器需要用弹性夹具固定,因为分散刀具在介质内高速旋转,容易产生巨大的吸力,极易导致玻璃容器破裂。11、如果仪器运转期间出现不平衡或异常噪声,应该立即停止,检查确认故障并更换分散刀具。12、不要长时间让分散刀具处于高速旋转状态,此时分散刀具和径向轴承可

[b][center]【第二届网络原创作品赛】药检之路:分散片检测解析过程[/center][/b][center][img]http://ng1.17img.cn/bbsfiles/images/2017/01/201701191651_625775_1612824_3.gif[/img][/center][center][img]http://ng1.17img.cn/bbsfiles/images/2009/08/200908240012_167324_1612824_3.jpg[/img][/center][b][color=#00008b][size=4]药检的同行们,你们平时都做哪些药物的检测?在检测中是否遇到过很棘手的问题呢?解析我们的检验,谈谈我们的看法,你今天的检测还都顺利吗?以下是我在工作中对片剂检测,还请大家多提宝贵意见,其中的错误与不足望老师专家斧正。[/size][/color][/b][color=#00008b][b]==================================================================================================================[/b][/color][color=#dc143c][size=3]罗红霉素分散片的检测[/size][/color][b]什么是分散片[/b]:系指在水中能迅速崩解并均匀分散的片剂。 分散片中的药物应是难溶性的。分散片可加水分散后口服,也可将分散片含于口中吮服或吞服。 分散片 应进行溶出度和分散均匀性检查。产品检测引用标准:《中华人民共和国药典》现行版技术标准:[img]http://ng1.17img.cn/bbsfiles/images/2009/08/200908240032_167326_1612824_3.jpg[/img]规格:75mg (每片含罗红霉素约75mg)有效期24个月(见包装封口)[b][color=#dc143c]********************************************************************************************************************[/color][/b][size=4][b][color=#dc143c]检验方法[/color][/b][/size][b]1.性状[/b]本品为白色或类白色[img]http://ng1.17img.cn/bbsfiles/images/2009/08/200908240038_167327_1612824_3.jpg[/img][color=#00008b]一般是目测,如发现变黄或缺损则判为不合格,同时假冒药片光泽性不好,易碎。[/color][b]2.鉴别[/b]在含量测定项下记录的色谱图中,供试品溶液主峰的保留时间应与对照品溶液的主峰的保留之间一致.[color=#00008b]可参照含量测定时色谱图(略)[/color][b]检查3.溶出度[/b][img]http://ng1.17img.cn/bbsfiles/images/2009/08/200908240046_167328_1612824_3.jpg[/img][color=#00008b]调试仪器到达试验要求,包括一些参数设定。[/color]测试前,调整装置,使转篮底部距溶出杯底部25mm±2mm.量取经脱气处理的盐酸溶液(1-1000)900ml为溶出介质,注入6个溶出杯中,加温使盐酸溶液温度保持在37±0.5度,转速为每分钟100转,并使其稳定.取本品6片,分别投入6个干燥的转篮内,将转篮降入容器中,立即开始计时,经30分钟时,取溶液适量,(取样点位置在转篮顶端至液面的中点,距溶出杯10mm处),立即经不大于0.8um的微孔滤膜过滤,自取样至过滤应在30秒完成,取续滤液作为供试溶液.另取本品10片,研细,精密秤取适量(相当于平均片重)加乙醇适量,(每5mg罗红霉素加乙醇1ml)使罗红霉素溶解,用溶出介质稀释100ml 过滤,取续滤液10ml加溶出介质80ml,制成每1ml约含80ug 罗红霉素溶液,作为对照.精密量取上述溶液各5ml,分别精密加入硫酸溶液(75-100)5ml,放置30分钟,冷却至室温,照”紫外分光光度计标准操作规程”在482nm的波长处分别测定吸光度,计算出每片的溶出度.应符合规定[img]http://ng1.17img.cn/bbsfiles/images/2009/08/200908240048_167329_1612824_3.jpg[/img][color=#00008b]注意取样时,取样的随机性,不能只在一盒药中取。[/color]计算含量A样:样品吸光度 A对:对照吸光度C对:对照溶液的浓度 g/mlW: 秤取的 供试品的量 gWo:为平均片重 g[img]http://ng1.17img.cn/bbsfiles/images/2009/08/200908240053_167330_1612824_3.jpg[/img][color=#00008b]注意取样时要迅速,并做好过滤处理,最好2人同时进行。在进行样品前处理时应避免样品二次污染。[/color][b]结果判定[/b]符合下列条件之一者,可判为符合规定(1)6片中,每片的溶出量,按标示量计算,均不低于82%,(2)6片中,如有1-2片低于82%,但不低于72%,且平均溶出量不低于82%(3)6片中,有1-2片低于82%,其中尽有一片低于72%,但不低于62%,且平均溶出量不低于82%时,应另取6片复试 初复试的12片中有1-3片低于82%,其中仅有1片低于72%,但不低于62%,且平均不低于82%.[img]http://ng1.17img.cn/bbsfiles/images/2009/08/200908240058_167331_1612824_3.jpg[/img][color=#00008b]紫外检测,认真填写检验记录,同时注意仪器使用和维护并做好记录。[/color]
求助,急啊!现在实验室要测定食品中的五氯硝基苯。 在测定食品中五氯硝基苯时,按照国标GB/T 5009.136-2003测定时,有一步骤说先在高速分散器中10000rpm分散4min,再2000rpm离心5min。 请问分散器和离心机有什么区别,你们实验室一般用什么型号的高速分散器? 另,国标GB/T 5009.136-2003规定的色谱柱为:内径3mm,长1.5m,内装涂1.5%OV+2%QF1固定液,担体为80~100目的 Chromsor WAW。 这里说的应该是填充柱吧?具体要用什么厂家什么型号的色谱柱好?换成Agilent 的DB-1701毛细管柱行不行?如果那位做食品农残的前辈能把你们公司有关有机氯农残检测的操作规程发给我一份,不胜感激,我的邮箱:xiaolou2003@126.com。
如题,有哪位同仁用过GC-MS检测致敏分散染料的,我正在寻求相关检测方法,请多多指教。
二极管阵列检测器做分散黄为什么是倒峰?请问有没有做过这个的
IKA T25分散机在农残检测中的使用与维护http://bbs.instrument.com.cn/shtml/20121220/4445080/
农药水分散粒剂的耐磨性检测仪器用哪种型号好
如题,检测水分散粒剂粒度范围的振筛机其振幅不超过5mm,振动次数应该为多少合适?哪位大虾知道,请指教一下,谢谢!

在生态纺织品检测中,有一种染料是分散黄39,有以下信息:CAS:12236-29-2,[url=https://insevent.instrument.com.cn/t/Yp][color=#3333ff]LC-MS[/color][/url]的谱图和标准物质的证书见下,求高手解析其结构:[img]http://ng1.17img.cn/bbsfiles/images/2009/05/200905271911_152277_1605875_3.gif[/img][img]http://ng1.17img.cn/bbsfiles/images/2009/05/200905271911_152278_1605875_3.gif[/img]其标准物质证书见下图[img]http://ng1.17img.cn/bbsfiles/images/2009/05/200905271914_152279_1605875_3.jpg[/img][img]http://ng1.17img.cn/bbsfiles/images/2009/05/200905271915_152280_1605875_3.jpg[/img]
有没有同学是做分散性染料的呢?有没有做分散性染料是用高效液相法(LC/DAD)分离检测二十几种染料的呢?有没有不带MS检测而只用LC/DAD检测分析的呢?求方法啊~
[size=4]请教各位,有没有关于水分散粒剂的分散性的检验标准,我们目前采用最后颠倒不超过10次为合格的标准,但我看有关水分散粒剂论文中,水分散粒剂分散性的检测数据都是百分之几十,我也查过有此方法,不知为何有两种方法。到底应该采用哪种为标准.[/size]
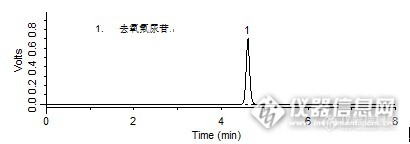
问题:去氧氟尿苷分散片:对照品溶液中理论塔板数是多少呢?答案:获奖名单:dahua1981(ID:dahua1981)吕梁山(ID:shih20j07)sixingxing(ID:v2889187)http://ng1.17img.cn/bbsfiles/images/2016/01/201601041759_580728_708_3.jpghttp://ng1.17img.cn/bbsfiles/images/2016/01/201601041759_580729_708_3.jpghttp://ng1.17img.cn/bbsfiles/images/2016/01/201601041800_580730_708_3.jpghttp://ng1.17img.cn/bbsfiles/images/2016/01/201601041800_580731_708_3.jpg【活动奖励】幸运奖(2钻石币):抽奖软件,当天随机抽取3个回答正确的版友ID号(最后一个ID号,截止至下午3:00),每人奖励2个钻石币积分奖励:所有回答正确的版友奖励10个积分(幸运奖获得者除外)。【注意事项】同样的答案,每人只能发一次PS:该贴浏览权限为“回贴仅作者和自己可见”,回复的版友仅能看到版主的题目及自己的回答内容,无法看到其他版友的回复内容。下午3点之后解除,即可看到正确答案、获奖情况及所有版友的回复内容。去氧氟尿苷分散片样品制备 制备方法含量测定对照品溶液:精密称取去氧氟尿苷对照品适量,加流动相使溶解并稀释制成每1 mL中含0.1 mg的溶液。分析条件 色谱柱Spursil C18 250 x 4.6 mm,5 μm (Cat#:82006)流动相水:乙腈:甲醇=75:5:20流速1.0 mL/min柱温30 ℃检测器UV 269 nm 进样量20 μL样品色谱图含量测定对照品溶液http://ng1.17img.cn/bbsfiles/images/2016/01/201601040958_580649_708_3.png 峰号 保留时间 min 峰面积 μV*s 峰高 μV 理论塔板数 N USP拖尾因子 分离度 1 4.610 4141058 703663 11157.732 1.125 -- *药典要求理论板数按去氧氟尿苷峰计算不低于1500本品种同时使用了DiamonsilC18色谱柱,在药典规定条件下进行检测,满足药典要求。
高效液相色谱法鉴别不同的分散染料*齐宝坤王景翰(中国刑警学院三系沈阳110035)提要用高效液相色谱法测定了100余种不同来源的分散染料。色谱柱为Novapak C18柱(150mm×3.9mmi.d. ,流动相为甲醇-水( 8∶ 15V/V) 检测波长为436nm 利546nm。关键词高效液相色谱法,分散染料1 前言不同厂家生产的分散染料,由于所用的原料和工艺不同,其产品的化学组成必有明显差异,利用这种差异可以进行鉴别。染料的测定方法有高效液相色谱法[1 2]、导数法[3]、正交函数法[4]等。本文用单纯形优化法选择流动相,系统地用高效液相色谱法鉴别了100余种分散染料,结果表明,用此方法可区分不同颜色和不同生产厂家牌号及相同牌号但生产厂家不同的分散染料。2 实验部分2.1 仪器和试剂Waters高效液相色谱仪,包括441型检测器,波长436nm和546nm,510型泵,U6K进样器和730型数据处理机;Novapak C18柱(150mm×3.9mm i.d. 。甲醇和N N-二甲基甲酰胺(DMF ,分析纯。二次蒸馏水。2.2 流动相的选择用单纯形优化的方法对甲醇和水二元因素进行调优。选择合适的初始顶点和步工及色谱优化函数,经过7次实验,确定最佳条件。流动相为甲醇∶水 85∶15琕V/V ,流速1.0mL/min。2.3 样品的制备和测定经用苯、二甲苯、氯仿和DMF的溶解试验表明,DMF对不同来源的分散染料溶解效果最好。故本文工作中均用DMF作染料溶剂,配制适当浓度的染玖料液,静置5 m n螅 取1 1 0 蘈重复3 次进 。色谱峰的保留时间和相对峰高取平均值。3 结果与讨论3.1 染料液测定结果我们共测定了100余种分散染料,将其中10种红、黄染料的测定结果列于表1。从表1中可以看出,由子不同颜色的染料化学结构截然不同,因此最大吸收波长、最强峰的tR值、峰数和相对峰高均不相同。表1 10种染料分析结果* 国家自然科学基金资助项目本文收稿日期:1995年1月2日,修回日期:1996年1月11日表1续)对于不同牌号相同颜色的染料,肉眼难以区分。但从表1中可以看出觯,5种红色 染料的峰迨 、⒏鞣峰tR值和相对峰高各不相同,黄色染料测定结果也均不相同,因此可以互相鉴别。这是因为这些染料成分和结构各异所致。本法也部可以区分同牌号不同生产厂家的染料。例如,A厂和B厂产的分散红3B在接近红色染料最大吸收波长546nm时,峰数都为4个,其中3个是弱峰,最强峰的tR都是5.23min,不易区分。但在波长为436nm时,它们的峰数和相对峰高值有明显差异,这是由于436nm接近黄色染料的最大吸收波长,这两种分散红3B虽然主成分相同,但杂质含量有差异。分散黄RGFL是生产厂家不同的同种染料,但在上述两个检测波长下,色谱峰数tR值和相对峰高也均不相同。3.2 重现性实验我们选择了两种分散红3B和两种分散黄RGFL染料,分别在两个波长下重复测定5次,相对标准偏差均在3.1和4.4以下,说明重现性较好。表2列谐出鯟厂产分散黄RGFL在546nm 波长下5次测定结果。表2 分散黄RGFL重现性实验参考文献1 齐宝坤,王景翰等.第十次全国色谱学术报告会文集,南京,19953412 Wheals.J Chromatogr,1985 350:2053 史晓凡,王景翰等.第二届微量物证检验学术文流会论文汇编,天津1990114 齐宝坤,史晓凡等.中国刑警学院学报,1994 1 28Discrimination of Various Disperse Dyes byHigh Performance Liquid Chromatography(HPLC)Qi Baokun and Wang Jinghan(Criminal Police College of China,Shenyang,110035)Abstract In this paper an HPLC method used to distinguish disperse dyes from different sources is presented.The operating conditions wereN ovapakC 18co lumn,150mm×3.9mm i.d.,MeOH/H2O(85∶15V/V)mobile phase and detection wavelength 436 and 546nm.The dye samples were dissolved in DMF and theninjected into HPLC column.By means of the values of absorption wavelength,peak number and relative peak height for each dye we candiscriminate about 100 disperse dye samples from different sources with satisfactory results.Key words high performance liquid chromatography,disperse dyes
题目:Comparison of the performance of conventional, temperature-controlled, and ultrasound-assisted ionic-liquid-dispersive, liquid-liquid microextraction combined with high-performance liquid chromatography in analyzing pyrethroid pesticides in honey samples摘要:This research paper presents a comparative study of the performance of conventional, ultrasound-assisted (UA), and temperature-controlled (TC) ionic liquid (IL) dispersive liquid-phase microextraction (IL-DLLME). Various parameters that affect extraction efficiency, such as type and volume of extraction and disperser solvent, centrifugation time, salt addition, effect of temperature on TC-IL-DLLME, and effect of sonication time on UA-IL-DLLME, were evaluated. UA-IL-DLLME was found to provide the best extraction efficiency. Under optimized conditions, great enrichment factors (506−515) and good recoveries (101.2%−103.0%) were obtained by analyte extraction in real samples. The limit of detections (LODs) ranged from 0.21−0.38 μg/L. Good linearity was obtained in the range of 0.5−200 µg L-1 for ethofenprox and tetramethrin, and 1−200 µg L-1 for meperfluthrin and alpha-cypermethrin. Based on optimized conditions, the UA-IL-DLLME method was applied and combined with high-performance liquid chromatography with diode array detection (HPLC-DAD) to determine the presence of ethofenprox, tetramethrin, meperfluthrin, and alpha-cypermethrin in honey samples.概述:对比了传统离子液体液液分散微萃取、温控辅助液液分散微萃取和超声辅助液液分散微萃取的区别,通过优化几个影响萃取实验的参数,如:萃取剂和分散剂的种类和体积、离心时间、盐效应、控温温度和超声时间等确定萃取条件,实验结果表明超声辅助液液分散微萃取的萃取效率要高于另外两种,在优化条件下四种菊酯类农药的富集倍数达到了506-515倍,在1−200 μg/L范围内具有良好的线性关系,线性相关系数在0.9990~0.9999 之间;检出限为0.04~0.10 μg/L(S/N=3),回收率为101.2%−103.0%。本方法已应用实际蜂蜜样本的检验,具有操作简单、富集效率高、灵敏度高和绿色环保等特点,可满足液体样品中菊酯类农药残留的检测要求。
发动机润滑油在发动机工作条件下,会产生多种污染物(包括氧化物、水分、金属颗粒、碳黑粒、酸、末完全燃烧物),这些污染物会使活塞表面覆盖一层漆膜。加有清净分散剂的润滑油可以阻止污染物粘结成团或粘结在金属表面上,抑制氧化反应,且能中和酸性氧化物,使污染物以溶胶状态分散地悬浮于油中,防止不溶物的沉积。这种性能的总和叫作发动机润滑油的清净分散性。 SH/T0645《柴油机油清浮性测定法(热管氧化法)》作为评定发动机润滑油清净性的手段之一。热管氧化试验是一种内燃机油高温氧化模拟台架试验设备,专门针对发动机活塞环等部件在工作过程中形成漆膜和积碳的机理而设计的试验方法。主要用于内燃机油高温清净性的实验室评定,考察油品中各类添加剂组分对油品的热氧化安定性、清净分散性等综合性能的影响。利用此类模拟试验技术可在进行IH2、IG2、IK等发动机台架试验之前,预先 筛选油品配方及评选各类添加剂的表现。试验测定的数据显示与台架试验结果有良好的相关性。SH/T 0300曲轴箱模拟试验法用于评定添加剂和含添加剂内燃机油的热氧化安定性,是科研工作中评选清净剂、抗氧抗腐剂和油品复合配方的一种模拟试验方法。该方法是使含添加剂内燃机油飞溅到高温金属表面形成漆膜,以此模拟曲轴箱油在活塞工作时的成漆情况,并用在试验机油箱内挂铅片的发放模拟曲轴箱油在气[url=https://insevent.instrument.com.cn/t/5p][color=#3333ff]液相[/color][/url]氧化状态下对发动机零部件的腐蚀。通过测定金属板上的漆膜评级和胶重,考察油的热氧化安定性。将250ml试样在规定条件下,在模拟试验机内运行6h后,考察形成漆膜和成胶的情况
从测试原理上理解,分散液(悬浮液)由于其颗粒的光散射作用,应该不能用吸光度测试其浓度。从有些版友的帖子里看到,粒度至少要小于100nm的分散液才能用吸光度测浓度。但曾看到国外有用吸光度测1微米粒径左右的分散液(一种分散染料);我同事也曾直接用水分散液测试,结果与使用有机溶剂将染料溶解后测试吻合得非常好。那么,究竟在什么条件下可以用吸光度测试分散液的浓度?
如题,现在非分散红外法基本都是将二氧化氮转换为一氧化氮再进行测量,但现在的转化炉效果都不理想,很多采用紫外差分直测法。请帮忙解释一下,为什么非分散红外测量NO2不稳定,而紫外差分却可以。
在分散染料标准DIN 54231-2005中是取0.5克样品加7.5ML甲醇萃取,然后上机测试。但是很多标准中限值是5mg/l 这个是怎么计算出来的呢?不应该是mg/kg才比较合理吗?
分散液液微萃取法原理是微量萃取剂在分散剂作用下形成分散的有机微液滴,均匀的分散与样品水溶液中,从而形成水/萃取剂/分散剂乳浊液体系,目标分析物被萃取到有机相中,最后在水样及微量萃取剂之间得到萃取平衡,最后离心分层。
我做分散的,请问,什么样的液体才算分散均匀了?比如我做的:能看到浑浊液,但是也有沉淀,分散均匀后的液体是不是应该是透明的啊?
请问各位老师,我用分散液液微萃取,二氯甲烷150ul为萃取剂,甲醇1ml为分散剂,涡旋之后只能取出70-80ul的二氯甲烷,老师说损失太多了。请问有什么方法减少这种损失吗?另外算富集倍率和萃取率体积按照70-80算呢还是150ul算呢?我加标回收率算出来在一百一左右。
请教下各位高手,我们公司是生产油漆的,所以用到颗粒状炭黑粉。但最近总是会出现炭黑不好分散堵机子,请问有没有什么方法在检测炭黑原材料的时候就能控制好呢?或是用什么方法能改善炭黑的分散性呢?谢谢!
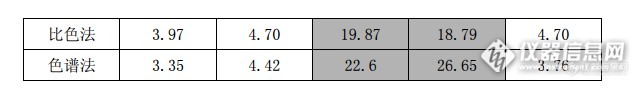
具体内容烦请查阅附件!http://simg.instrument.com.cn/bbs/images/default/emyc1002.gif分散液相微萃取分析法分析废水探讨【引言】 随着苯胺装置产能的不断提升,有机废水处理也相应成为稳定生产中很重要的一环。目前采用的比色法普遍存在操作繁琐、持续时间长(全程 5 小时左右)、难以实现自动化等缺点,经常出现废水处理发生异常后得不到及时有效的控制,以致影响生化系统的正常运行。因此,建立快速、准确的废水分析方法是非常必要的,为突发的生产异常事件在最短的时间得出分析结果,以便异常能够得到及时的处理。【摘要】 分散液相微萃取是最近发展起来的一种新型样品前处理技术,只需要不超过 2ml的有机溶剂,环境友好,成本低廉、富集效率高,预处理过程及其简单,单个样品预处理可以在 3 分钟完成,该方法与气相色谱联用分析废水,单个样品出结果时间能够控制在 30 分钟以内。本方案针对我公司废水的实际情况,对样品体积、萃取剂和分散剂的选择,萃取剂使用体积、萃取时间的确定以及影响样品富集的盐度、酸度、温度等方面加以评述。1、分散液相微萃取技术的原理 分散液相微萃取类似于液液萃取,是基于目标分析物在样品溶液和小体积的萃取剂之间平衡分配的过程。而对于具有酸碱性的分析物,可通过控制样品溶液的 pH 值使分析物以非离子化状态存在,从而提高分配系数。在样品溶液中加入萃取剂和分散剂,混合液经轻轻振荡后即形成一个水/分散剂/萃取剂的乳浊液体系,再经离心分层,用微量进样器取出萃取剂就直接进样分析。该方法集采样、萃取和浓缩于一体,能够有效提高样品分析物的回收率和富集倍数。2、萃取剂的种类及体积的影响 萃取剂需满足两个条件:一是其密度必须大于水,这样才能通过离心的方法把水溶液与萃取剂分离;二是萃取剂要不溶于水而且对待测物的溶解能力要大,以保证取得良好的萃取效率,萃取剂的选择是保障待测组分较高回收率的重要因素。本方案选用氯化苯、氯仿、二氯甲烷、二硫化碳、四氯化碳做比对试验,充分考虑萃取剂对硝基苯和苯胺的萃取效率的影响,并使离心之后的沉积基本控制在固定体积之内,以消除沉积相体积不同对萃取结果造成的影响,由于事故池废水浓度较高,普遍高达上万个 ppm,容易造成样品超出样品的富集极限,造成回收率降低;同时还要兼顾总排口、二沉池废水浓度低,富集倍数需要提高的现状,故沉积体积也不适宜太大,经过反复比较,将沉积体积控制在 80μl 左右能够兼顾本公司各类废水样品的回收与富集。介于目前我们公司多数废水同时含有硝基苯很苯胺的现实,使用二硫化碳和二氯甲烷进行联合萃取,微量的氯化苯能够促使样品的乳化程度,促使待测组分与萃取剂有较大机会的接触,使待测组分得到较好的提取和富集,同时,也能得到较好的色谱峰形。3、分散剂的选择及体积的影响 分散剂的选择是影响萃取效率的另一个关键因素,分散剂可以使萃取剂在水相中分散成细小的液滴,均匀的分散在溶液中,即形成一个水/分散剂/萃取剂的乳浊液体系,增大萃取剂与待测物的接触面积,从而提高萃取效率。分散剂(丙酮)体积的变化必然导致沉积相体积的改变。在沉积相体积基本相同的条件下考察萃取剂体积对萃取效率的影响。萃取回收率的变化趋势表明,随着丙酮体积的增加,萃取回收率先增加继而减少。在低体积的丙酮中, 萃取剂分散得不好, 传质效果差,导致萃取回收率降低。随着丙酮体积增大,苯胺在水中的溶解性增强,萃取效率下降。硝基苯影响不大,经比较:选择1.5 mL的丙酮为最佳条件。4、萃取和离心分离时间的确定 萃取时间是影响待测组分回收率的一个重要因素。在分散液相微萃取中,萃取时间是指在水相中注入了萃取剂和分散剂后,到混合液开始离心之前这段时间。研究表明,萃取时间对DLLME萃取效率没有显著的影响,这是由于在溶液形成乳浊液之后萃取剂被均匀地分散在水相中,待测物可以迅速由水相转移到有机相并达到两相平衡。萃取时间短是分散液相微萃取的一个突出的优点。试验表明:离心2分钟能够达到理论沉积体积的要求,故确定离心时间为2分钟,离心速度为4000r/h。5、样品溶液体积的影响 在萃取剂/分散剂体积一定的情况下,改变样品溶液的体积,分别取2.5、5、和10mL为考察体积。结果表明,在样品溶液为2.5 mL时,萃取剂过饱和,分离后沉积明显减少,所得到富集效率不能达到最大值;当样品溶液为5 mL时,萃取剂在样品溶液中达到基本饱和,此时富集效率达到相对最佳值;继续增大样品溶液的体积至10、15 mL时,所得萃取效率明显下降,尤其不适应事故池和原水样品组分的分离 ,表明样品溶液过量,导致样品在水相和萃取剂之间不能有较好的平衡,从而使富集倍数降低。实验选用样品溶液的最佳体积为5 mL。6、萃取温度的影响 考虑到实验中萃取剂、分散剂和样品溶液的混合可能存在微弱的热交换,实验表明该热交换不足以对萃取效率产生影响。但萃取时环境温度影响沉积相的生成,对萃取效率有一定的影响。随着萃取时温度的升高,沉积相体积逐渐减少,环境温度达到 50℃时,胶束体系基本消失,沉积相体积几乎降至 0,故合适的萃取温度范围为 5~20 ℃,实验在温度 20 ℃下进行,对于汽提塔废水需要降至室温后才可以施行样品预处理。7、盐浓度的影响 随着离子强度的增加,分析物和有机萃取剂在水相中的溶解度减小,利于提高回收率,所得到的沉积体积也有所增加,公司内各类废水都经过 PH 调节等处理,故不需要考虑盐度对萃取效率的影响。8、PH 的影响 酸性条件下对于废水中硝基苯的提取影响不大,在 PH 为 2-12 的范围内,回收率仍然不会受太大的影响,而废水体系中的苯胺在 PH 低于 7 时,回收率急剧下滑,酸度降至 3 左右时,回收率将跌至 10%以下,故废水预处理前的 PH 不容忽视,提取水相中苯胺的最佳酸度范围为 9-109、重复性试验 本试验方案在含量为 1-500ppm 时具有最好的重复性,当浓度高于 1000ppm 后重复性逐渐下降。【试验部分】DLLME 法样品处理过程相对比较简便,只需经过加样、萃取、分离三步骤即完成样品的预处理,全过程耗时不超过 5 分钟。1、试剂与仪器1.1 卤苯(CP)1.2 二氯甲烷(CP)1.3 二硫化碳(CP)1.4 丙酮(CP)1.5 气相色谱仪(FID 检测器)1.6 高速离心机1.7 移液枪(1000-5000μL)2、试验方法2.1 色谱条件 DB-1701 毛细管柱(60m×0.3mm×0.25μm),起始柱温 110 ℃,保持 3 min,以 10 ℃/min 的速率升温至 220 ℃,并保持 10 min;载气为高纯 N2,流速 30 mL/min;尾吹气流速 60 mL/min;采用分流进样,分流比 10∶1。进样口温度 250 ℃;检测器温度为270 ℃。2.2 分散液液微萃取步骤 取 5 mL 澄清样品溶液于离心试管中,缓慢加入含萃取剂的丙酮溶液 1.5mL。轻轻振荡离心管,使样品溶液和丙酮溶液形成胶束体系,将离心管放入离心机内,调节离心机转速为 4000 r/min,离心 2 min,用微型进样器取 1μL,进气相色谱分析。【综合评估】1、DLLME-GC 在高浓度废水中的应用目前有机类废水硝、胺基类含量分析方法系引用 GB11889 N-(1-萘基)乙二胺偶氮分光光度法,该方法最高监测上限为 1.6mg/L,该方法硝基酚对结果成正干扰,浓度高的废水理论上可以通过稀释来获取近似真实值的结果,然而高倍数的稀释所带来的误差非常惊人,无法指导生产,以下将引用 7 月 8 日苯胺厂冷冻
分散液相微萃取分析法分析废水探讨张燕【新浦化学(泰兴)有限公司】【引言】随着苯胺装置产能的不断提升,有机废水处理也相应成为稳定生产中很重要的一环。目前采用的比色法普遍存在操作繁琐、持续时间长(全程5小时左右)、难以实现自动化等缺点,经常出现废水处理发生异常后得不到及时有效的控制,以致影响生化系统的正常运行。因此,建立快速、准确的废水分析方法是非常必要的,为突发的生产异常事件在最短的时间得出分析结果,以便异常能够得到及时的处理。【摘要】分散液相微萃取是最近发展起来的一种新型样品前处理技术,只需要不超过2ml的有机溶剂,环境友好,成本低廉、富集效率高,预处理过程及其简单,单个样品预处理可以在3分钟完成,该方法与气相色谱联用分析废水,单个样品出结果时间能够控制在30分钟以内。本方案针对我公司废水的实际情况,对样品体积、萃取剂和分散剂的选择,萃取剂使用体积、萃取时间的确定以及影响样品富集的盐度、酸度、温度等方面加以评述。1、分散液相微萃取技术的原理分散液相微萃取类似于液液萃取,是基于目标分析物在样品溶液和小体积的萃取剂之间平衡分配的过程。而对于具有酸碱性的分析物,可通过控制样品溶液的pH值使分析物以非离子化状态存在,从而提高分配系数。在样品溶液中加入萃取剂和分散剂,混合液经轻轻振荡后即形成一个水/分散剂/萃取剂的乳浊液体系,再经离心分层,用微量进样器取出萃取剂就直接进样分析。该方法集采样、萃取和浓缩于一体,能够有效提高样品分析物的回收率和富集倍数。2、萃取剂的种类及体积的影响萃取剂需满足两个条件:一是其密度必须大于水,这样才能通过离心的方法把水溶液与萃取剂分离;二是萃取剂要不溶于水而且对待测物的溶解能力要大,以保证取得良好的萃取效率,萃取剂的选择是保障待测组分较高回收率的重要因素。本方案选用氯化苯、氯仿、二氯甲烷、二硫化碳、四氯化碳做比对试验,充分考虑萃取剂对硝基苯和苯胺的萃取效率的影响,并使离心之后的沉积基本控制在固定体积之内,以消除沉积相体积不同对萃取结果造成的影响,由于事故池废水浓度较高,普遍高达上万个ppm,容易造成样品超出样品的富集极限,造成回收率降低;同时还要兼顾总排口、二沉池废水浓度低,富集倍数需要提高的现状,故沉积体积也不适宜太大,经过反复比较,将沉积体积控制在80μl左右能够兼顾本公司各类废水样品的回收与富集。介于目前我们公司多数废水同时含有硝基苯很苯胺的现实,使用二硫化碳和二氯甲烷进行联合萃取,微量的氯化苯能够促使样品的乳化程度,促使待测组分与萃取剂有较大机会的接触,使待测组分得到较好的提取和富集,同时,也能得到较好的色谱峰形。3、分散剂的选择及体积的影响分散剂的选择是影响萃取效率的另一个关键因素,分散剂可以使萃取剂在水相中分散成细小的液滴,均匀的分散在溶液中,即形成一个水/分散剂/萃取剂的乳浊液体系,增大萃取剂与待测物的接触面积,从而提高萃取效率。分散剂(丙酮)体积的变化必然导致沉积相体积的改变。在沉积相体积基本相同的条件下考察萃取剂体积对萃取效率的影响。萃取回收率的变化趋势表明,随着丙酮体积的增加,萃取回收率先增加继而减少。在低体积的丙酮中, 萃取剂分散得不好, 传质效果差,导致萃取回收率降低。随着丙酮体积增大,苯胺在水中的溶解性增强,萃取效率下降。硝基苯影响不大,经比较:选择1.5 mL 的丙酮为最佳条件。4、萃取和离心分离时间的确定萃取时间是影响待测组分回收率的一个重要因素。在分散液相微萃取中,萃取时间是指在水相中注入了萃取剂和分散剂后,到混合液开始离心之前这段时间。研究表明,萃取时间对DLLME萃取效率没有显著的影响,这是由于在溶液形成乳浊液之后萃取剂被均匀地分散在水相中,待测物可以迅速由水相转移到有机相并达到两相平衡。萃取时间短是分散液相微萃取的一个突出的优点。试验表明:离心2分钟能够达到理论沉积体积的要求,故确定离心时间为2分钟,离心速度为4000r/h。5、样品溶液体积的影响 在萃取剂/分散剂体积一定的情况下,改变样品溶液的体积,分别取2.5、5、和10 mL为考察体积。结果表明,在样品溶液为2.5 mL时,萃取剂过饱和,分离后沉积明显减少,所得到富集效率不能达到最大值;当样品溶液为5 mL时,萃取剂在样品溶液中达到基本饱和,此时富集效率达到相对最佳值;继续增大样品溶液的体积至10、15 mL时,所得萃取效率明显下降,尤其不适应事故池和原水样品组分的分离,表明样品溶液过量,导致样品在水相和萃取剂之间不能有较好的平衡,从而使富集倍数降低。实验选用样品溶液的最佳体积为5 mL。6、萃取温度的影响 考虑到实验中萃取剂、分散剂和样品溶液的混合可能存在微弱的热交换,实验表明该热交换不足以对萃取效率产生影响。但萃取时环境温度影响沉积相的生成,对萃取效率有一定的影响。随着萃取时温度的升高,沉积相体积逐渐减少,环境温度达到50℃时,胶束体系基本消失,沉积相体积几乎降至0,故合适的萃取温度范围为5~20 ℃,实验在温度20 ℃下进行,对于汽提塔废水需要降至室温后才可以施行样品预处理。7、盐浓度的影响随着离子强度的增加,分析物和有机萃取剂在水相中的溶解度减小,利于提高回收率,所得到的沉积体积也有所增加,公司内各类废水都经过PH调节等处理,故不需要考虑盐度对萃取效率的影响。8、PH的影响酸性条件下对于废水中硝基苯的提取影响不大,在PH为2-12的范围内,回收率仍然不会受太大的影响,而废水体系中的苯胺在PH低于7时,回收率急剧下滑,酸度降至3左右时,回收率将跌至10%以下,故废水预处理前的PH不容忽视,提取水相中苯胺的最佳酸度范围为9-109、重复性试验本试验方案在含量为1-500ppm时具有最好的重复性,当浓度高于1000ppm后重复性逐渐下降。【试验部分】DLLME法样品处理过程相对比较简便,只需经过加样、萃取、分离三步骤即完成样品的预处理,全过程耗时不超过5分钟。1、试剂与仪器1.1卤苯(CP)1.2 二氯甲烷(CP)1.3二硫化碳(CP)1.4 丙酮(CP)1.5 气相色谱仪(FID检测器)1.6 高速离心机1.7 移液枪(1000-5000μL)2、试验方法2.1色谱条件 DB-1701毛细管柱(60m×0.3mm×0.25μm),起始柱温110 ℃,保持3 min,以10 ℃/min的速率升温至220 ℃,并保持10 min;载气为高纯N2,流速30 mL/min;尾吹气流速60 mL/min;采用分流进样,分流比 10∶1。进样口温度250 ℃;检测器温度为270 ℃。2.2 分散液液微萃取步骤取5 mL澄清样品溶液于离心试管中,缓慢加入含萃取剂的丙酮溶液1.5mL。轻轻振荡离心管,使样







 我要推广仪器
我要推广仪器
 下载APP
下载APP