岛津GC-2014C,检测是没有峰值出现,点火能够点着,可是显示检测器没有控制
[color=#444444]我之前用恒温测了单个物质的浓度,并且做了标准曲线(浓度 vs. pA),现在我要测多个物质混合物,需要程序升温,其他流量,进样量都不变,只改变进样口温度,柱温和检测器温度,这样同物质同浓度出来的峰值和我恒温测出的峰值一样吗?这条标准曲线还可以继续使用吗?如果能用我就不用再做单个物质的程序升温了,好多好慢好费时间。急求,多谢!~[/color]
请教各位大神:甲苯和醋酸仲丁酯混合后经检测为何只出一个峰值? 甲苯和醋酸仲丁酯单独进样出峰时间都是一样的!后将载气流速调慢,结果是一样的!请问怎样才能将这两种物质区分开来?
液相色谱检测(被测物是纯度比较高的)制成一定系列浓度,再用液相色谱检测,是不是进样浓度越高,峰值就越大
高校液相色谱测四溴双酚A,测的值峰值减少很多,减少至少10倍,为什么?测的峰值积分面积比之前测的小了许多,是不是样品的PH会有影响?
在hplc的使用过程中,同一种样品的检测,峰值越来越小,我想问一下是否是检测器的寿命问题。一般石墨电极的寿命是多少个小时?还是柱子的问题,柱子的平均寿命是多少?谢谢
求教各位版友~对这台机器还是比较菜,求大家帮忙~~着急~~GCMS 6890-5973,做测量审核样品——大米粉中的毒死蜱含量测定使用内标法定量,在制作标准曲线时,各点的内标含量均为0.5ppm(毒死蜱标准品标明用丙酮稀释,内标为之前储存也,接手前就一直使用丙酮稀释)样品按照国标处理,加内标亦为0.5ppm,最终用正己烷定容1mL标准曲线点跑出的谱图峰值很低,内标峰值为500-,而样品所得谱图内标峰值为1100+怕是基质问题,做过如下标准品①将购买的毒死蜱标准品用丙酮稀释,加内标,用正己烷定容1mL②将购买的毒死蜱标准品用丙酮稀释,加内标,用丙酮定容1mL结果相同~由于大米粉中毒死蜱含量未知所以只能确定内标的峰值过低,现在谱图不方便放上来,晚些时候来放图,期限又要到了,着急T.T求大家帮忙~~谢谢!!~万分感激!!~========================================================================2013.11.13 关于此次出现问题的最终解决方法反馈:2013.11.08检测结果已经寄送出去,11.11被对方签收,目前结果还没有出来,出后会补发的~现只总结下该问题的最终解决办法~~虽然具体原因不清楚,不过希望写出来能够有点帮助~~测过几次都出现同样问题后,另一位同事根据她测苏丹红的经验,猜测是不是应该把配置好的标准品也过下柱子于是将标准品配置好后过柱子备用,同时防止再出现问题,更换了新的衬管标准品与样品上机后,两者内标峰值基本保持一个水平了~约为30,000+标准曲线R值较好,回收率试验三次,平均回收率为104%以上就是基本的情况哦~~换衬管后也有侧过没过柱子的标准品,与换衬管前相似所以,为什么过了柱子后峰值变大,原因不明不知大家有何高见~~
我做火焰法测铅,用2,4,6,8,10ppm梯度时,样品溶液检测不出;后来用0.04,0.08,0.12,0.16ppm梯度可以得到一条r=0.997的线,样品测出大约1.2ppm左右。问题是后一次梯度下ABS值非常小,峰值不明显,这样的曲线还能要么?[em40]
刚接触蛋白分离纯化,现在在过柱子分离纯化蛋白。现在过的是sephadex G50设备是 凝胶柱 紫外检测仪 部分收集器 不能在电脑上显示峰图。我一直有个困惑,紫外检测器检测到蛋白时显示屏上数值会上升,上升反应的是蛋白浓度逐渐升高吗?随着数值升高 我该如何收集峰值处的蛋白呢 ,我是5ml/管收集。 举例子:数值从1到30一个试管 5ml过去了 第二个试管中数值45(峰值),正常我应该收集第二试管的对吧? 那第一个试管里有我要的蛋白吗? 到底怎么收集?求大家帮我解答一下 真的不明白 搞不懂!
在给一个学生侧样品时,他样品袋子上写的AlN 但是测得的峰值有二三十万 用软件查了一下也没查出是什么东西来 请问什么东西的峰值有这么高啊 [em0904]
经XRD衍射仪检测的样品图谱,在HighScore Plus进行图谱分析时,衍射峰的峰尖上如何标注相应的元素,匹配后的图谱有几百种候选项该如何进一步确定具体的物象,求大神指教!
KJ-08W型流动注射氢化物发生器,预喷0,积分5s,读出的峰值只有屏幕上显示最大值的一半多点,但是能成线性。岛津6300默认是读峰高还是峰面积?如何才能读到峰值?我是哨音响后即读数。 哨音向后读数,迟两三秒,修改积分时间都试过,都读不到峰值。咋个办?
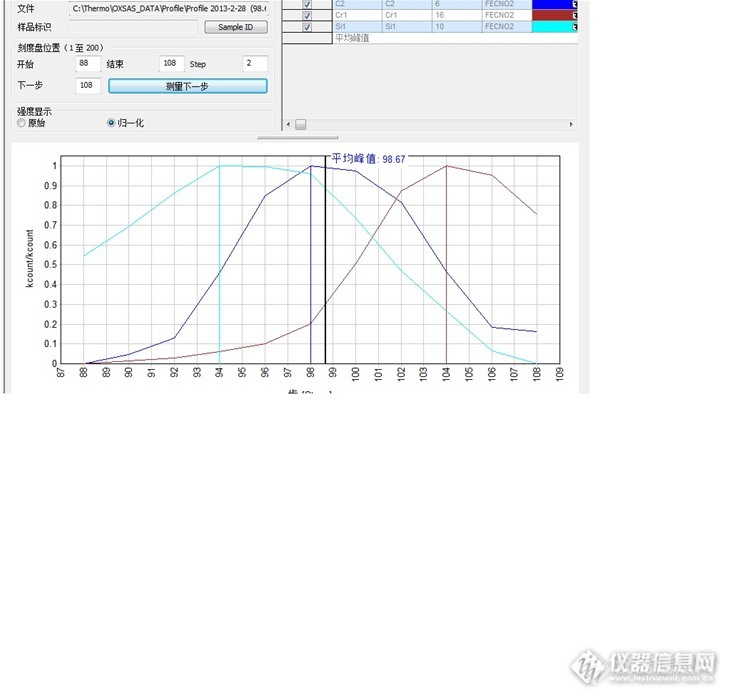
今天按照维护计划更换了真空泵油,按照维护手册的方法,更换真空泵油后要对设备进行积分描迹。但是描迹完后看峰值有些疑惑。见图:1、设备刚买后进行的描迹,时间在2011年4月http://ng1.17img.cn/bbsfiles/images/2013/02/201302282108_427682_1617441_3.jpg2今晚刚完成的描迹http://ng1.17img.cn/bbsfiles/images/2013/02/201302282110_427684_1617441_3.jpg看图后,谁能给解释下为什么现在的几个峰值相差距离很大啊,而当时几乎都在一块的,都是98的。相差间距大对设备描迹取峰值有影响吗,能否代表现在的是光通过最大的位置。是否需要重新选取几个接近的代表通道呢?今天早上重新选择标样进行描迹,几个峰值以在一个点上,确定是原来标样选择错误,希望大家吸取教训,不要犯同类型错误了。
如题:求助1,2,3-三唑(http://ng1.17img.cn/bbsfiles/images/2013/12/201312041240_480686_2834894_3.gif)的红外峰值,特征峰值
用原子荧光测硒,仪器型号为瑞利610A,1ppm的Se(4+),没有峰值。用相同条件,测砷都有信号的。是为什么啊?是是灯的问题了,还是浓度太大了?谢谢各位
我在外校做了单晶MgO的激光拉曼图,但没有及时核对标准谱图,希望朋友们手头有手册的,帮我查一下它的拉曼峰值区。谢谢
由于我处于一个图书短缺的大学中,在外校做了几个锰、镍、钴、钛的无机材料的激光拉曼图,但没有及时核对标准谱图,现在写文章,希望朋友们手头有手册的,帮我查一下上面几个元素在3价、2价氧化态的拉曼峰值区。谢谢
我做的注射用胞磷胆碱钠成品含量和有关物质时候,由于对照品干燥后放置时间过长有3个小时左右做出来结果不合,第二次对照品干燥后马上实验,发现对照品峰值有明显不同,放置时间长的有明显下降,对照品(140675-200803)储存条件是遮光阴凉干燥处,用前要105℃干燥4小时,是否降解,请高人帮忙看看
请教高手,做红外,空白用KBr压片,显示的图谱按“计算”的按钮,会自动检出峰值标示在图谱上,然后接着扫描样品片,出现图谱之后按“计算”的按钮,没有峰值标示在图谱上???为什么呢|???
测试仪器ICP6300 方法:基体匹配,测试杂质Pb测试结果,空白(加基体)峰值118 0.2ppm铅的标液(加基体)峰值125 1ppm铅的标液(加基体)峰值149这样可以用吗还有我拿空白当样品测,测试值0.1PPM, RSD 12拿0.2ppm标液当样品测,测试值0.27ppm RSD平均16拿1ppm标淮测试,测试值0。95ppm RSD 6(不知道是不是RSD,就是那个反映重复的指标)我想问的是这样能拿来测样品吗,,,我测了一下是0。3ppm RSD 17凝问:空白拿来测怎么不是零的 一致性怎么这么差

[img]http://ng1.17img.cn/bbsfiles/images/2009/03/200903071706_137216_1631661_3.jpg[/img]小弟接触红外时间不久,请教大家一个问题QQ:57536786E-mail:sunzhimems@163.com愿听赐教!图是我的薄膜样品,分别从薄膜的二个面入射而做出来的红外图。简单看来他们的峰数基本一致,但是在指纹区他们的峰值相差了大约一半啊?教科书上说:峰值与跃迁几率的大小和振动偶极距变化的大小有关。跃迁几率大和振动偶极距变化大则吸收峰强。那么对与我做的同一种材料的二个侧面,他们的峰值相差一半,应该怎么解释一下那?! 还是由于跃迁几率,偶极距变化吗? 二个侧面的成分含量不一样吗?!
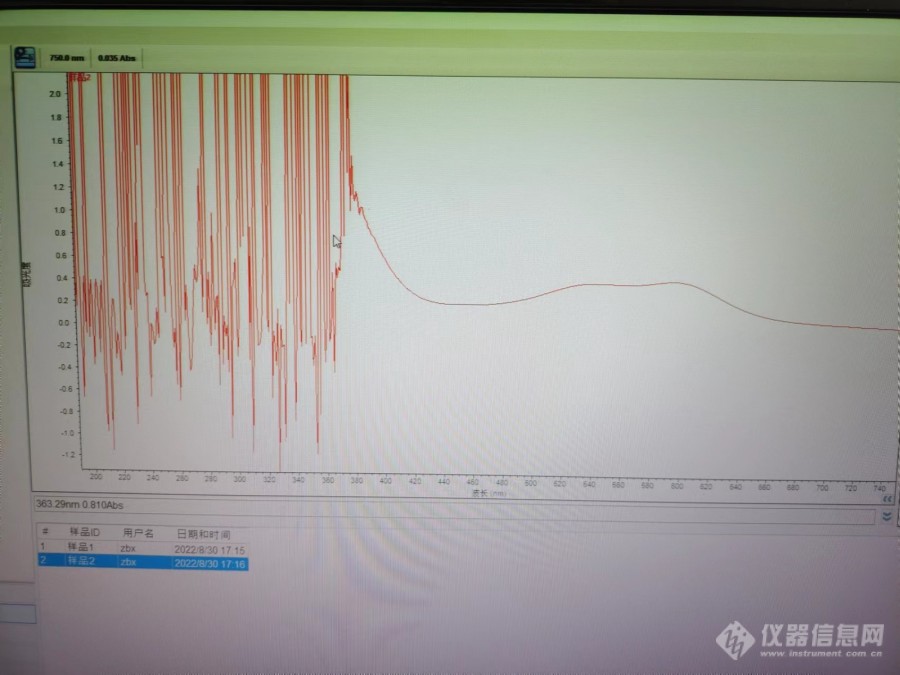
本人是新手,在做香草醛--高氯酸法测皂苷,用乙酸乙酯做稀释剂测全波长扫描紫外区域峰值变化乱,且可见光区域特征峰不明显。而用冰醋酸做稀释剂可见光区域有明显的峰值。请问一下大家,紫外区域混乱的峰值是怎么回事?在一个稀释剂冰醋酸和乙酸乙酯对峰值的影响?第一个图为标品(齐墩果酸)的扫描图,稀释剂为乙酸乙酯。第二个图为样品扫描图,稀释剂为冰醋酸。第三个图为样品全波长扫描,稀释剂为乙酸乙酯。[img=标准品400-700扫描(乙酸乙酯稀释剂),690,517]https://ng1.17img.cn/bbsfiles/images/2022/09/202209071149091261_5045_5794499_3.jpg!w690x517.jpg[/img][img=样品400-750波长(冰醋酸稀释剂),690,517]https://ng1.17img.cn/bbsfiles/images/2022/09/202209071149514409_994_5794499_3.jpg!w690x517.jpg[/img][img=样品全波长(乙酸乙酯稀释剂),690,517]https://ng1.17img.cn/bbsfiles/images/2022/09/202209071148091671_6988_5794499_3.jpg!w690x517.jpg[/img]
请教,同样的物质同样的条件测出来的DTA的峰值是不是比DSC的峰值温度低啊?有没有文献支持的啊?
峰值不对原因
我用岛津LC-20A测定完物质后,报告定制时定义了峰值,可是拖入数据后看不到峰值的情况,请问是怎么回事啊?
用液相做黄酮实验,峰值越跑越低是什么原因呢?就是同一样品,第一针峰值高,从第二针还是越跑越低?请教,求解。
请问,无机颗粒填充环氧树脂复合材料的DMA图谱中,损耗角正切值的高度代表阻尼行为,那么填料经过表面处理,参与环氧树脂交联体系,那么损耗角正切峰值高度是增高还是降低?我的理解是:体系交联度增大,刚性增强,阻尼减小,损耗角正切峰值高度应该降低;但是,另外一个角度考虑,界面作用力增大,内摩擦增大,阻尼行为应该增大啊(即损耗角正切峰值高度增高)请大家看看哪种理解是对的?
我是个新手,刚接手[url=https://insevent.instrument.com.cn/t/3p][color=#3333ff]离子色谱[/color][/url]不久。早上[url=https://insevent.instrument.com.cn/t/3p][color=#3333ff]离子色谱[/color][/url]进不了样,六通阀堵了,好不容易自己拆了六通阀解决以后,把仪器安装好重新测水样的氟离子浓度。结果做水样有峰值,但是数据却显示n.a.。这个是什么原因啊?样品的保留时间,标样是2min多一些,但是样品却是3min多一些,这又应该怎么处理呢?求求大家帮帮我吧刚出来工作就遇到这种事好慌啊
Rid-10a 用的75:25的乙腈水作为流动相,柱子是氨基柱 先进系统冲柱子冲平了后进了一个空针(图1)就很平没有峰值,然后点系统里检测器按钮冲了大概两三个小时还是不平,就进了一个空针看见了很多峰值(图2)然后点了平衡检测器冲了会儿点的零点,就开始进了一针样品针(图3)出现图二一样的杂峰,求助!各位小伙伴们有没有遇到过这种问题怎么解决啊
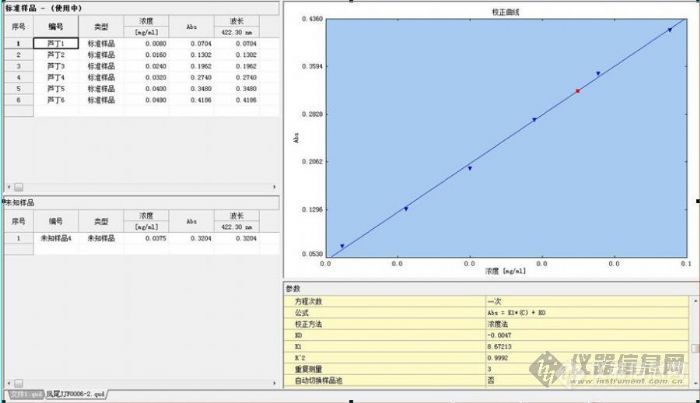
这个问题我有一点困惑,前两天别人送来一样品叫我帮他做个紫外定量,标准液是芦丁。我在设定量参数时,就问他们芦丁的最大吸收波长是多少?他们查了资料是410nm,我就建议他们让我先帮他们先做一个光谱扫描寻找最大吸收波长,然后我选用了芦丁1(0.008mg/ml)标液进行光谱扫描,结果峰太平,峰值检测结果是418.7nm,我觉得用最低浓度找最大吸收波长不可靠,于是我就让他们换成最高浓度芦丁6(0.049mg/ml)标液进行光谱扫描,结果峰形满意,峰值检测结果是422.3nm(可惜忙忘了保存,否则可传给大家看),然后再设定量参数就用了422.3nm,做出的定量结果可看附件上的图,结果好的让我意外,R值也达到了三个9,送样者也很满意。 我把结果放在这里晒,希望大家解我困惑: 1、以上我测样过程有哪些不对的地方? 2、在怀疑文献上的最大吸收波长时,选择标液中哪一个浓度进行光谱扫描比较合适、科学?[img]http://ng1.17img.cn/bbsfiles/images/2007/12/200712082139_72254_1644065_3.jpg[/img]







 我要推广仪器
我要推广仪器
 下载APP
下载APP