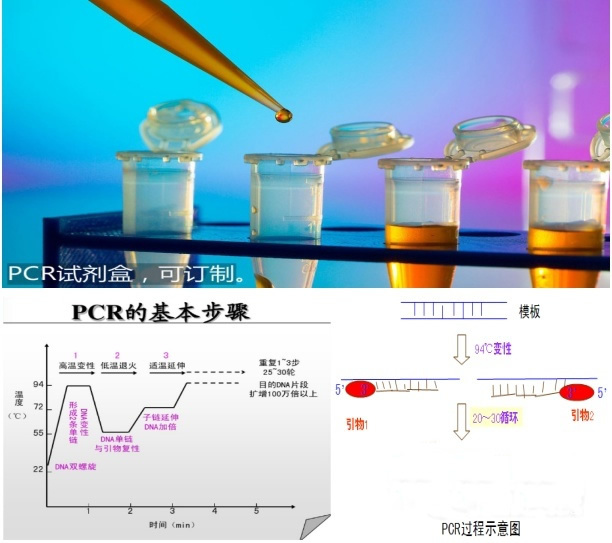
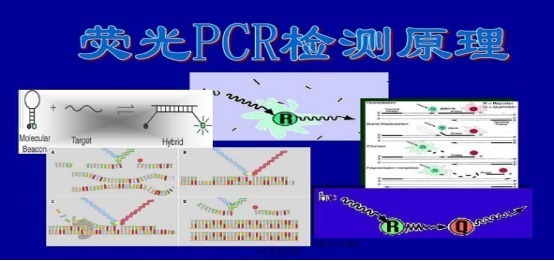
推荐厂家
暂无
暂无




 400-629-8889
400-629-8889
 留言咨询
留言咨询
 400-873-7896
留言咨询
400-873-7896
留言咨询
 400-860-8560
留言咨询
400-860-8560
留言咨询



近日,中科院广州生物医药与健康研究院曾令文研究组成功开发出一种基于核酸等温链置换反应技术、T4连接酶反应与胶体金技术的单核苷酸多态性检测的生物传感器。 该生物传感具有以下三个特点:1.简单,无需复杂的检测仪器,仅需室温反应;2.高灵敏度,单次反应可检测约6个核酸分子;3.高特异性,可区分单碱基突变。该生物传感器克服了传统检测方法的操作技术复杂、耗时长、需要特殊仪器等缺陷。 该生物传感器可用于检测/诊断由单核苷酸多态性引起的遗传病、耐药性病原微生物、肿瘤等疾病易感基因。相关成果于7月6日发表在国际著名学术期刊Chemical Communications,2012,48(68): 8547-8549。 该项目由国家重大专项(2008ZX10004-004)、(2009ZX1004-109)经费资助。http://www.cas.cn/ky/kyjz/201208/W020120830533626771243.jpg单核苷酸多态性检测生物传感器
人类基因组单核苷酸多态性的研究进展与动态The research development of single nucleotide polymorphisms in human genome 摘要:第一张人类基因组序列草图已经公布,正式图预计也将于2003年4月完成。但序列图只基于少数个体,它反映了基因组稳定的一面,并未反映其变异或多态的一面,而正是这种多态性,即基因组序列的差异构成了不同个体与群体对疾病的易感性、对药物与环境因子不同反应的遗传学基础。人类基因组中存在广泛的多态性,最简单的多态形式是发生在基因组中的单个核苷酸的替代,即单核苷酸多态性(single nucleotide polymorphisms, SNPs)。SNP通常是一种二等位基因的(biallelic),即二态的遗传变异,在CG序列上出现最为频繁。在转录序列上的SNP称为cSNP。SNP的数量大、分布广。按照1%的频率估计,在人类基因组中每100~300个核苷酸就有一个SNP。因此,整个人类基因组(3.2 X 109bp)中至少有1,100万以上的SNPs,在任何已知或未知基因内和附近都可能找到数量不等的SNP 目前普遍认为,作为数量最多且易于批量检测的多态标记,SNP在连锁分析与基因定位,包括复杂疾病的基因定位、关联分析、个体和群体对环境致病因子与药物的易感性研究中将发挥愈来愈重要的作用。迄今,对多基因疾病候选基因的SNPs研究已积累了丰富的数据,基于这些SNPs的关联分析也正方兴未艾。本文阐述了SNP的特征、不同研究者对基于SNP进行关联分析的观点以及SNP的研究进展与动态。 关键词: SNP;遗传标记;关联研究 中图分类号:Q75 随着分子遗传学的进展,疾病遗传学研究从简单的单基因疾病转向于复杂的多基因疾病(如骨质疏松症、糖尿病、心血管疾病、精神性紊乱、各种肿瘤等)与药物基因组学的研究中。与前者相比,多基因性状或遗传病的形成,受许多对微效加性基因作用,即其中每种基因的作用相对较微弱。这些不同基因构成的遗传背景中,可能有易感性主基因(major gene)起着重要作用。它们同时还受环境因素的制约,彼此间相互作用错综复杂,所以任一基因的多态性对疾病发生仅起微弱的作用。鉴于此,需要在人类基因组中找到一种数目多、分布广泛且相对稳定的遗传标记,单核苷酸多态性(single nucleotide polymorphisms, SNPs)正是代表了这样一种标记,所以它成为继第一代限制性片段长度的多态性标记、第二代微卫星即简单的串联重复标记后,第三代基因遗传标记。 1. SNP作为遗传标记的优势 SNP自身的特性决定了它比其它两类多态标记更适合于对复杂性状与疾病的遗传解剖以及基于群体的基因识别等方面的研究。 (1)SNP数量多,分布广泛。据估计,人类基因组中每1000个核苷酸就有一个SNP,人类30亿碱基中共有300万以上的SNPs。SNP 遍布于整个人类基因组中,根据SNP在基因中的位置,可分为基因编码区SNPs(Coding-region SNPs,cSNPs)、基因周边SNPs(Perigenic SNPs,pSNPs)以及基因间SNPs(Intergenic SNPs,iSNPs)等三类。 (2)SNP适于快速、规模化筛查。组成DNA的碱基虽然有4种,但SNP一般只有两种碱基组成,所以它是一种二态的标记,即二等位基因(biallelic)。 由于SNP的二态性,非此即彼,在基因组筛选中SNPs往往只需+/-的分析,而不用分析片段的长度,这就利于发展自动化技术筛选或检测SNPs。主要的技术方法包括单链构象多态性(single strand conformation polymorphisms, SSCPs)法、异源双链分析(heteroduplex analysis, HA)、DNA直接测序分析、变异检测阵列(variant detector arrays, VDA)法以及基质辅助激光解吸附电离飞行时间(MALDI-TOF)质谱法等。 (3)SNP等位基因频率的容易估计。采用混和样本估算等位基因的频率是种高效快速的策略。该策略的原理是:首先选择参考样本制作标准曲线,然后将待测的混和样本与标准曲线进行比较,根据所得信号的比例确定混和样本中各种等位基因的频率。 (4)易于基因分型。SNPs 的二态性,也有利于对其进行基因分型。对SNP进行基因分型包括三方面的内容:(1)鉴别基因型所采用的化学反应,常用的技术手段包括:DNA分子杂交、引物延伸、等位基因特异的寡核苷酸连接反应、侧翼探针切割反应以及基于这些方法的变通技术;(2)完成这些化学反应所采用的模式,包括液相反应、固相支持物上进行的反应以及二者皆有的反应。(3)化学反应结束后,需要应用生物技术系统检测反应结果。目前许多生物技术公司发展出高通量检测SNP的技术系统,如荧光微阵列系统(Affymetrix)、荧光磁珠技术(Luminex,Illumina, Q-dot)、自动酶联免疫(ELISA)试验(Orchid Biocomputer)、焦磷酸的荧光检测(Pyrosequencing)、荧光共振能量转移(FRET)(Third Wave Technologies)以及质谱检测技术(Rapigene, Sequenom)。 2. 基于SNP的关联研究 如果某一因素可增加某种疾病的发生风险,即与正常对照人群相比,该因素在疾病人群中的频率较高,此时就认为该因素与疾病相关联。如非遗传因素吸烟与肺癌相关;在遗传因素中,如APOE4与Alzheimer`s相关。对疾病进行关联分析需要在年龄与种族相匹配的患者和对照人群中确定待测因素(环境的或遗传的)的频率分布,患者和对照人群的选择是否恰当直接影响结果的可靠性。对常见的由高频率、低风险等位基因导致的疾病,采用致病等位基因的关联分析比连锁分析更有效。 应用SNP进行关联研究,首先需明确多少SNPs才可满足在全基因组范围内的分析。Kruglyak应用计算机模拟法预测人类基因组中超过3Kb就不存在连锁不平衡,据此推出完成全基因组扫描将需要500,000个SNPs。而Collins等收集通过家系研究得到的常染色体单倍型的信息发现,在染色体上相距0.2cM到0.4cM(约200-400kb)之间的标记仍存在连锁不平衡,如按每100kb需要一个SNP计算,那么完成全基因组扫描仅需约30,000个SNPs,平均每3-4个基因用一个SNP就可识别出整个基因组内任何位置上的具表型活性的变异。最近发现SNP与SNP之间的连锁不平衡甚至可延伸到更远的区域(0.35cM-0.45cM),那么进行基因组扫描需要的SNP数量就更少。导致上述估算SNP 数量差异的主要原因是Kruglyak进行模拟计算时,假设现在的人群在5000年前起源于共同的祖先,且人群规模的有效大小保持在10,000左右,然后经过连续的指数扩增,直至达到现在的50亿左右。Collins认为这种假设是不现实的,在人类发展的历史过程中,人群数目的增长是迂回曲折的,经历扩张与萎缩的周期性变化。 Weiss等认为Collins及其同事的结果可能低估了问题的复杂性。因为他们的结果或是基于小样本资料推断出来的,就会使连锁不平衡(LD)程度的估算偏高;或是从理论上预测LD的水平,而忽略了基因组中大量的随机变异。如大多数位点的信息是来源于小样本中测序得到的资料,据此得到的单倍型结构不可靠。目前的研究集中于基因组中LD相对广泛存在的区域,在此区域内,基因相对容易作图。如基于这些经验来进行基因组其它区域的LD分析,就可能发生偏离。如两个相距较远的SNPs 之间具有强的LD性质,就认为它们之间的SNPs及该SNP侧翼的SNPs也存在强烈的LD,这种假设仅适合于其中一些多态位点,但它并不是通则。当然,在一些罕见人群中,如Saami,在较长的区域内广泛存在大量的LD,但对Fihland人群,则在较长区域内几乎不存在LD,对全球整个复杂人群而言,LD肯定变得更复杂一些。 Gray等认为随着人类基因组测序计划的进展,人类基因组的结构逐渐被阐明,因此就可在那些富含基因的区域选择SNP进行全基因组扫描,这样所需的SNP数量还会减少。Halushka等根据他们对75个基因检测的实验结果推测,SNPs在单个基因或整个基因组中的分布是不均匀的,在非转录序列中要多于转录序列,而且在转录区也是非同义突变的频率比其它方式突变的频率低得多。Templeton 等对LPL基因突变与重组热点的研究结果提示,SNP集中分布于基因组的CG二核苷酸处或单核苷酸重复区或αDNA聚合酶的识别位点(TGGA)处。将人类基因组不同区域物理图谱与遗传图谱的进行比较,发现遗传距离和物理距离的比值有很大的差异,提示基因组不同区域的重组水平存在差异。如Dunham等将22号染色体STR的物理位置与遗传位置进行了对比,发现该染色体的重组率差异很大,提示存在重组热点。根据基因组内不同区域重组频率的高低可进一步选择SNP的数量,重组热点需要的标记数量就多,相反就少。这种设计也可能会进一步减少基因组扫描所需的SNP标记。 使用SNP进行关联分析面临的另一个问题是如何选择SNP。如果对每一个SNP都进行独立研究,那么对几百万SNPs 的研究就会导致成千上万次的假关联,结果就掩盖真实的关联性,所以,进行关联分析前,一定要对所研究的SNP进行选

[size=3][font=宋体]是指在基因组上单个核苷酸的变异[/font][font=Times New Roman],[/font][font=宋体]包括置换、颠换、缺失和插入。从理论上来看每一个[/font][font=Times New Roman]SNP [/font][font=宋体]位点都可以有[/font][font=Times New Roman]4 [/font][font=宋体]种不同的变异形式[/font][font=Times New Roman],[/font][font=宋体]但实际上发生的只有两种[/font][font=Times New Roman],[/font][font=宋体]即转换和颠换[/font][font=Times New Roman],[/font][font=宋体]二者之比为[/font][font=Times New Roman]2 :1[/font][font=宋体]。[/font][font=Times New Roman]SNP [/font][font=宋体]在[/font][font=Times New Roman]CG[/font][font=宋体]序列上出现最为频繁[/font][font=Times New Roman],[/font][font=宋体]而且多是[/font][font=Times New Roman]C[/font][font=宋体]转换为[/font][font=Times New Roman]T ,[/font][font=宋体]原因是[/font][font=Times New Roman]CG[/font][font=宋体]中的[/font][font=Times New Roman]C [/font][font=宋体]常为甲基化的[/font][font=Times New Roman],[/font][font=宋体]自发地脱氨后即成为胸腺嘧啶。一般而言[/font][font=Times New Roman],SNP [/font][font=宋体]是指变异频率大于[/font][font=Times New Roman]1 %[/font][font=宋体]的单核苷酸变异。在人类基因组中大概每[/font][font=Times New Roman]1000 [/font][font=宋体]个碱基就有一个[/font][font=Times New Roman]SNP ,[/font][font=宋体]人类基因组上的[/font][font=Times New Roman]SNP [/font][font=宋体]总量大概是[/font][font=Times New Roman]3 ×10[sup]6 [/sup][/font][font=宋体]个[/font][font=Times New Roman] [/font][font=宋体]。[/font][/size][size=3][font=Times New Roman] [/font][/size][size=3][font=宋体]因此[/font][font=Times New Roman],SNP[/font][font=宋体]成为第三代遗传标志[/font][font=Times New Roman],[/font][font=宋体]人体许多表型差异、对药物或疾病的易感性等等都可能与[/font][font=Times New Roman]SNP[/font][font=宋体]有关。[/font][/size][size=3][font=宋体] 现在普遍认为[/font][font=Times New Roman]SNP[/font][font=宋体]研究[/font][font=宋体]是人类基因组计划走向应用的重要步骤。这主要是因为[/font][font=Times New Roman]SNP[/font][font=宋体]将提供一个强有力的工具,用于高危群体的发现、疾病相关基因的鉴定、药物的设计和测试以及生物学的基础研究等。[/font][font=Times New Roman]SNP[/font][font=宋体]在基因组中分布相当广泛,近来的研究表明在人类基因组中每[/font][font=Times New Roman]300[/font][font=宋体]碱基对就出现一次。大量存在的[/font][font=Times New Roman]SNP[/font][font=宋体]位点,使人们有机会发现与各种疾病,包括肿瘤相关的基因组突变;从实验操作来看,通过[/font][font=Times New Roman]SNP[/font][font=宋体]发现疾病相关基因突变要比通过家系来得容易;有些[/font][font=Times New Roman]SNP[/font][font=宋体]并不直接导致疾病基因的表达,但由于它与某些疾病基因相邻,而成为重要的标记。[/font][font=Times New Roman]SNP[/font][font=宋体]在基础研究中也发挥了巨大的作用,近年来对[/font][font=Times New Roman]Y[/font][font=宋体]染色体[/font][font=Times New Roman]SNP[/font][font=宋体]的分析,使得在人类进化、人类种群的演化和迁徙领域取得了一系列重要成果。[/font][/size]









 我要推广仪器
我要推广仪器
 下载APP
下载APP