推荐厂家
暂无
暂无




 400-628-5299
400-628-5299
 留言咨询
留言咨询
 400-628-5299
留言咨询
400-628-5299
留言咨询
 400-860-5168转2735
留言咨询
400-860-5168转2735
留言咨询



建立运用RP-HPLC-PDA测定灯盏花素分散片中灯盏花乙素的方法。色谱条件为:色谱柱:C18 柱(250 mm×4.6 mm,5μm);流动相:甲醇:0.1%磷酸(V∶V=40∶60);流速:1.0 mL/min;紫外检测波长:335nm;柱温:40℃。测定的线性范围:0.39~3.9μg (R= 0.9999, n=7);加标回收率:97%~99%;方法精密度(RSD): 1.97%。
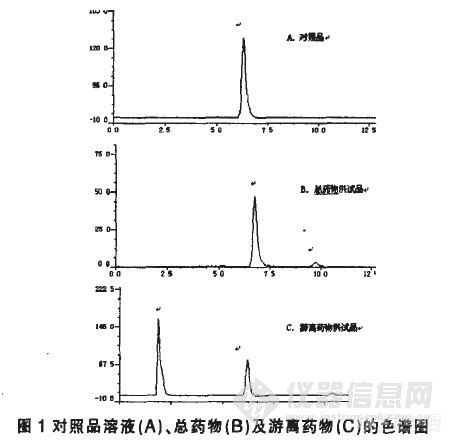
作者:李文秀;邓英杰;魏伟;高晓非;曹金娜(沈阳药科大学药学院 沈阳药科大学药学院 沈阳药科大学药学院 沈阳药科大学药学院 沈阳药科大学药学院)摘要:目的:建立灯盏花素前体脂质体胶囊的突释考察方法。方法:采用透析-高效液相色谱法(HPLC),水化前体脂质体后,用透析袋分析游离药物和脂质体,并用HPLC法测定含量。采用Diamonsil ODS C18色谱柱(4.6mm×250mm,5μm),流动相为甲醇-乙腈-40mmol/L KH2PO4(磷酸调pH至2.5)(18∶12∶70);检测波长335nm;流速1.0ml/min;柱温35℃。结果:游离药物在透析膜两侧均匀分布,而脂质体被截留于透析袋中。含量测定方法平均加样回收率为103.32%,RSD为1.12%(n=9)。结论:本法操作简便、准确,可用于灯盏花素前体脂质体胶囊突释的评价。谱图:http://ng1.17img.cn/bbsfiles/images/2012/08/201208211652_385078_1609970_3.jpg

“虐”我两天的灯盏花前言 最近实验室来了一批灯盏花素原料药,实验室几个同事做了好几次含量测定都不符合要求。上级领导吩咐让我接手,展开实验室内部调查。于是乎一场和灯盏花素的“较量”正式开始了。 我所使用的主要的仪器有:Agilent 1260 --VWD 液相色谱仪 赛多利斯 BS-125D 电子分析天平 首先从检验的标准依据开始,打开药典灯盏花素的页面。如下图所示:http://ng1.17img.cn/bbsfiles/images/2014/09/201409252107_515688_2204446_3.jpg 刚开始感觉这个方法很普通,就是一个简单的液相色谱方法,没这么在意。按部就班的配制流动相,称取对照品和样品稀释超声出理等一系列的过程忙活起来,两个小时后样品盒对照品都处理好了,上机测试开始,打第一针后问题就来了。 第一个问题是理论踏板数不符合要求。标准要求不得低于5000,(下图中红圈部分,明确要求要达到5000)。http://ng1.17img.cn/bbsfiles/images/2014/09/201409252127_515690_2204446_3.jpg 而我们的测定结果只有3000多。没办法我找来了三根色谱柱开始,选一个符合要求的使用。三根柱子分别编号1, 2,3号。http://ng1.17img.cn/bbsfiles/images/2014/09/201409252123_515689_2204446_3.jpg 使用1号柱子的色谱图如下:http://ng1.17img.cn/bbsfiles/images/2014/09/201409252132_515691_2204446_3.jpg很明显1号柱子的色谱峰严重拖尾,不能用来定量。只好更换第2号柱子分析。分析结果如下:http://ng1.17img.cn/bbsfiles/images/2014/09/201409252136_515693_2204446_3.jpg由上图可知色谱峰的峰型大有改善,峰很好看,关键的是理论塔板数只有 3576,仍然不能满足5000的要求。只好使用3号根住子,3号是150mm的短柱,分析结果如下:http://ng1.17img.cn/bbsfiles/images/2014/09/201409252145_515697_2204446_3.jpg由3号柱子的分析结果可以看出,色谱峰的峰型较2号柱的峰型胖了点。峰宽由0.1985变成了0.4723,保留时间有5.043min变成了14.956min.而理论塔板数由3576,变成了5555。终于符合了标准系统适应性理论塔板数不低于5000的要求。苦逼的自己,忙活了一整个下午终于把测定条件搞定了,抬头一看早已经过了下班的时间了,急急忙忙把仪器序列运行上。期待第二天的测定结果。 第二天一上班,就赶紧处理昨天晚上分析结果,结果除掉水分的含量只有88.5%。还是低于90%的标准要求。检测结果还是不是很满意,上报领导检测结果。领导指示看看是不是前处理出问题了,于是一个细节性的问题出来了,药典样品前处理中明确要求要是用300W,50KHz的超声波超声处理45分钟。而我们的超声波清洗器的超声频率只300W,40KHz,超声的频率不够吗?样品没有超好,主要成分没有完全溶解吗?马上行动,更换一台300W,50KHz的超声处理样品和照品溶液。上机分析测试,匆匆忙忙大半天又过去了,好在吃完午饭后,分析结果终于出来了。除掉水分后的含量还是只有88.4%。确认检测结果没问题后,赶紧上报领导,领导看完结果后,很不情愿的说:“下不合格报告吧”。我终于可以松口气了。 总结 本次试验的整个过程,难点在于系统适应性理论塔板数的要求。影响塔板数的因素有两个,一是保留时间,另一个是峰宽。他们之间的关系是:塔板数与保留时间成正比,与峰宽成反比。所以保留时间越长,塔板数越大,峰宽越小塔板数越大。 三号柱是短柱保留时间应该更短,而三号柱的保留时间却变长了,原因是我改变了流动相的比例,增加了水相的比例。 值得我们探讨的是为什么很多中药的检测塔板数要求都很高,按照标准的流动相很难满足要求的?如我的峰型很好, 塔板数达不到标准要求,会影响到检测结果的准确性吗?流动相的比例调节的范围一般是多少?










 我要推广仪器
我要推广仪器
 下载APP
下载APP