推荐厂家
暂无
暂无
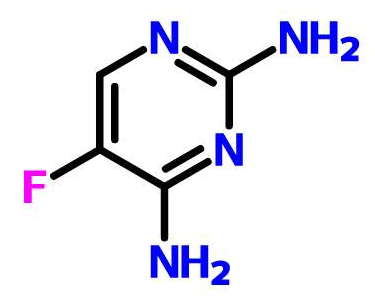
有一个亚胺类化合物(标样)用甲醇和水作流动相,出现了两个色谱峰,而用乙腈和水却只有一个峰。请问各位这是为什么?难道是甲醇与亚胺反应?谢谢!
化合物纯度的鉴定方法,从快速,便宜,简便的要求出发,主要来之于以下几点:一 通过TLC的纯度的鉴定, 我将自己的心得分述如下1 展开溶剂的选择,不只是至少需要3种不同极性展开系统展开,我的经验是首先要选择三种分子间作用力不同的溶剂系统,如氯仿\甲醇,环己烷\乙酸乙酯,正丁醇\醋酸\水,分别展开来确定组分是否为单一斑点.这样做的好处是很明显的,通过组份间的各种差别将组分分开,有可能几个相似组份在一种溶剂系统中是单一斑点,因为该溶剂系统与这几个组分的分子间力作用无显著的差别,不足以在TLC区分.而换了分子间作用力不同的另一溶剂系统,就有可能分开.这是用3种不同极性展开系统展开所不能达到的.2 对于一种溶剂系统正如wxw0825所言,至少需要3种不同极性展开系统展开,一种极性的展开系统将目标组分的Rf推至0.5,另两种极性的展开系统将目标组分的Rf推至0.8,0.2。其作用是检查有没有极性比目标组分更大或更小的杂质。3 显色方法,光展开是不够的,还要用各种显色方法。一般一定要使用通用型显色剂,如10%硫酸,碘,因为每种显色剂(不论是通用型显色剂,还是专属显色剂在工作中都遇到他们都有一化合物不显色的时候),再根据组分可能含有混杂组份的情况,选用专属显色剂。只有在多个显色剂下均为单一斑点,这时才能下结论样品为薄层纯二 通过熔程,判断纯度。原理很简单,纯化合物,熔程很短,1,2度。混合物熔点下降,熔程变长。三,基于HPLC的纯度鉴定,对于HPLC因为常用的系统较少,加之其分离效果好,我们一般不要求选择三种分子间作用力不同的溶剂系统,只要求选这三种不同极性的溶剂系统,使目标峰在不同的保留时间出峰。四,基于软电离质谱的纯度鉴定。如ESI-MS,APCI-MS。大极性化合物选用ESI-MS,极性很小的化合物选用APCI-MS,这些软电离质谱的特点是只给出化合物的准分子离子峰,通过正负离子的相互沟通来确定分子量。如果样品不纯,就会检出多对准分子离子峰,不但确定了纯度,还能明确混杂物的分子量。五,基于核磁共振的纯度鉴定,从氢谱中如果发现有很多积分不到一的小峰,就有可能是样品是样品中的杂质。利用门控去偶的技术通过对碳谱的定量也能实现纯度鉴定。好了,不能再多写了。这里只是对常见的纯度鉴定方法做了一个小结,从快速,便宜,简便的要求出发,以第一点最合要求,往后次之,所以对第一点详加讲述。当然每种方法多有各自的局限性,如基于氢谱的纯度鉴定,如果发现有很多积分不到一的小峰,还有可能使样品中的活泼质子,基于软电离质谱的纯度鉴定,如果混杂物的分子量与目标物一样就无法检出。等等还有很多。这需要大家在工做中积累,思考。要讲的话,我看好几篇都讲不完。最后说一下对化合物纯度的要求,世界上不存在100%纯的化合物。你希望要多高的纯度应该与你的目的有关,例如,如想测核磁共振鉴定结构,一般要求95%的纯度,如果想测EI-MS,纯度越高越好。99%以上。还有,以上的方法都不能区分对应异构体。
[b]手性(chirality)[/b]是三维物体的基本属性,三维结构的物体所具有的与其镜像的平面形状完全一致,但在三维空间中不能完全重叠的性质,正如人的左右手之间的关系。具有手性的化合物即称为[b]手性化合物[/b],手性化合物除了通常所说的含手性中心的化合物外,还包括含有手性轴、手性平面、手性螺旋等因素的化合物。一般来说,如果分子既无对称面也无对称中心,分子就具有手性。手性分子绝对构型的确定是一个极其重要且长期存在的问题。目前确定手性分子绝对构型的方法主要有四类:(1) 有机化学法;(2) 核磁共振法;(3) X射线衍射法;(4) 光谱法,如旋光光谱法、圆二色谱、振动圆二色谱等。[b]1. 有机化学法[/b]有机合成是最早的确定分子手性的方法,主要为化学相关法。即将目标分子反合成分析,从初始已知手性的化合物开始,通过手性控制的有机化学反应,将其转化为目标化合物的方法,然后从他们旋光符号或者相应的[url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]、液相色谱推导出其绝对构型。很多富有挑战性的复杂手性化合物的合成如今已被有机化学家们所攻克,然而有机合成始终是一项繁琐而辛苦的选择。[color=#000000][b]2. 核磁共振法(NMR)[/b][/color][color=#000000]NMR 技术是获的化合物结构的首选方法,其耦合常数和NOE谱图是获取化合物相对构型的重要手段,适用于刚性结构非对映体的构型确认。但是对于光学(对映)异构体而言,一般情况下其NMR谱的信号是相同的,即应用NMR 谱无法直接将其区分,也不能确定其绝对构型。近年来发展了一些间接方法,借助NMR法,通过手性样品的衍生物来测定对映异构体的绝对构型。[/color][color=#000000]在应用NMR法测定手性化合物绝对构型的方法中,以Mosher 法最为常用。即通过将样品衍生化为非对映异构体或类似于非对映体,测定样品分子与手性试剂反应后产物的[sup]1[/sup]H-NMR 或[sup]13[/sup]C-NMR 位移数据,得到其化学位移的差值并与模型比较,最后推定底物分子手性中心的绝对构型。例如,Mosher法是将待测样品的手性仲醇基(或仲胺基)与([i]R[/i])或([i]S[/i])-α-甲氧基-α-三氟甲基-α-苯基乙酸(亦称Mosher 酸,缩写MTPA,见图1)反应生成相应的酯或酰胺,然后测定该酯或酰胺的核磁共振氢谱。根据MTPA芳香环的屏蔽效应,比较待测物与MTPA成酯(或酰胺)前后[sup]1[/sup]H-NMR 或[sup]13[/sup]C-NMR信号的化学位移差,由谱中化学位移的差值和模型图来推测仲醇(或仲胺基)的绝对构型。[/color][align=left]手性衍生物的NMR法的样品用量少,衍生物合成简单,测定迅速、准确,在手性醇、手性胺、手性羧酸的绝对构型确定中已经非常成熟。由于目前所开发的手性识别剂主要针对于手性中心中的某些基团(如羟基、氨基、羧酸),并且需要昂贵的手性试剂进行衍生化,其应用范围有所局限。 [/align][color=#000000][b]3. X-射线衍射法(X-raydiffraction)[/b][/color][color=#000000]普通的X-射线法(钼靶)仅能构筑化合物的相对构型,不能区分对应异构体。如果分子中含有重原子(一般原子序数大于16)或在分子中引入一个重原子,就可用X-射线来测定该重原子的手性分子绝对构型。此外,通过引入另一个已知绝对构型的手性分子也可获得结构的绝对构型。随着技术的发展,采用CuKa作为入射光源的X-射线单晶CCD衍射仪,对于测定相对分子量在1000以下、含C、H、N、O原子有机分子的绝对构型已可实现了。[/color][color=#000000]在单晶结构分析中,目前国际公认表征绝对构型的参数称为Flack 参数,当结构分析进入到最后的精修阶段时,如果该参数等于或接近0,或其参数在± 0.3之内,那么一般认为绝对构型就被确定了。[/color][color=#000000]采用单晶X-衍射法样品用量少、测定迅速、结果可靠直观,可以作为最终的立体构型的确定方法。但是由于测试的仪器价格昂贵,对单晶有严格要求,也限制了X-射线衍射法的应用。[/color][color=#000000][b]4. 光谱法[/b][/color][color=#000000]在光谱分析方法中,现有最有名和应用最广泛的手性分子构型确定法为旋光光谱法(ORD) 和圆二色谱法 (CD),该法对样品要求不高 (如纯度、官能团、结晶等)、测量过程无损失,因而得到了广泛应用。近几年,振动圆二色谱法 (VCD)取得了巨大的发展,逐渐成为一项鉴定手性分子绝对构型的重要工具。[/color][color=#000000][b]4.1 旋光光谱法(ORD)[/b][/color][color=#000000]早期的手性光学法是旋光谱法。当平面偏振光通过手性物质时, 能使其偏振面发生旋转,这种现象称之为旋光。 用仪器记录通过手性化合物溶液的平面偏振光的振动面偏转的角度,即为旋光度α,我们平常所测定的旋光即为波长在589.6 nm的Na灯的黄光下的比旋光度。旋光度随波长的变化而变化就可获得旋光光谱(ORD)。[/color][color=#000000]在同系物中,相同的化学反应使旋光值按相同的方向改变,而不改变其旋光的方向,因此通过比较相关化合物的旋光性,可得到手性化合物的构型信息。在采用该方法测定药物绝对构型时,应与绝对构型已知且与待测药物结构相同或相似化合物,在相同的实验条件下测定旋光光谱,以保证比较结果的可靠性。[/color][color=#000000]相比圆二色谱法(CD)而言,CD谱形尖锐、简单明了、易于分析,ORD现已被现代手性光学技术CD所取代。[/color][color=#000000][b]4.2 圆二色谱法 (CD)[/b][/color][color=#000000]传统的圆二色谱所用的平面偏振光的波长范围一般在紫外区(200~400 nm)。手性化合物(溶液)在左旋和右旋圆偏振光的吸收系数(ε)之差随入射偏振光波长的改变而改变, 得到的图谱即是圆二色光谱(CD),又称为电子圆二色谱(ECD)。[/color][color=#000000]该方法主要是通过测定光学活性物质(待测物)在圆偏振光下的Cotton效应,根据Cotton效应的符号获得药物结构中发色团周围环境的立体化学信息,并与一个绝对构型已知的与待测药物结构相似化合物的Cotton效应相比较,或者借助计算化学的方法,对比实验测值和理论计算值,即可能推导出待测物的绝对构型。[/color][color=#000000]长期以来,电子圆二色谱由于其干扰少、容易测定而被广泛应用。但该法使用的前提条件是待测化合物的手性中心含有合适的发色团(有紫外吸收),或者能够引进合适的发色团。对于手性中心无发色团或无法引入发色团的化合物,则不适宜采用该方法。[/color][color=#000000][b]4.3 振动圆二色谱法 (VCD)[/b][/color][color=#000000]传统的圆二色谱要求手性分子必须有紫外吸收,这一点成为限制其应用的重大问题。在20世纪70年代,Holzwart,Nafie和Stephens等先后成功测定了红外光区频率下的圆二色谱,即振动圆二色谱(VCD)。当平面偏振光的波长范围在红外区(4000~750 cm-1)时,由于其吸收光谱是分子的振动转动能级跃迁引起的,VCD谱即为红外光中的左旋圆偏光和右旋圆偏光的吸收系数之差∆ ε随波长变化所给出的图谱。[/color][color=#000000] 由于振动光谱谱图的复杂性, VCD很难象传统圆二色谱 (electronic circular dichroism, ECD)那样发展出合适的理论来进行结构-谱图的对应解释,主要依靠理论计算值和实测值对比来判断手性分子的绝对构型。[/color][color=#000000]与ECD相比,VCD的最大优势就是不需要分子中含有生色团 (紫外吸收),几乎所有手性分子都在红外区有吸收,都会产生VCD谱图。此外,VCD测试是在溶液状态测定,不需要单晶,样品中的非手性杂质也不影响测定结果。随着越来越多的关注和研究,振动圆二色谱法将成为一项鉴定手性分子绝对构型的强有力的工具。[/color][color=#000000]除上述的四大经典构型确定法外,红外光谱、紫外光谱法也用于辅助测定化合物的构型。更多的方法还望同行们共同探讨总结,希望大家在讨论区多多给予意见,谢谢![/color]本帖摘自“手性专家”微信公众号。





 400-889-7796
400-889-7796
 留言咨询
留言咨询
 400-877-2799
留言咨询
400-877-2799
留言咨询
 400-805-8969
留言咨询
400-805-8969
留言咨询









 我要推广仪器
我要推广仪器
 下载APP
下载APP