推荐厂家
暂无
暂无
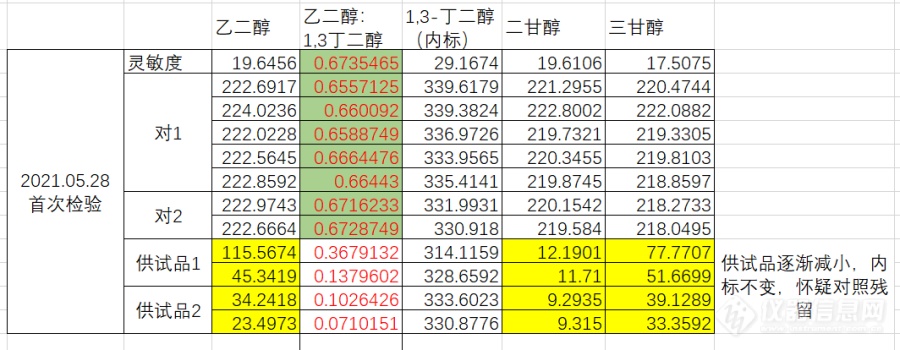
在进行聚山梨酯80Ⅱ 乙二醇、二甘醇和三甘醇检测过程中,发现对照品溶液重复性相当不错,但是供试品中目标峰逐渐减小,见如下数据:[img=第一次实验时实验数据,690,268]https://ng1.17img.cn/bbsfiles/images/2021/07/202107031131180784_3076_3356999_3.png!w690x268.jpg[/img]当时出现这种情况,便怀疑是对照品残留到了供试品中,然后减少了进样针数,实验数据如下:[img=减少对照进样次数,690,147]https://ng1.17img.cn/bbsfiles/images/2021/07/202107031133386103_879_3356999_3.png!w690x147.jpg[/img]供试品确实小了很多,而且也没有逐渐减小的情况,当时基本确定是对照品残留,因此后续实验调整了进样顺序,首先进供试品,但是又出现跟首次检验一样的情况,供试品目标峰面积逐渐减小,而后面的对照重复性也是相当不错做到此时怀疑是色谱柱问题,因此新采购了色谱柱,但供试品逐渐减小的问题仍然存在,后续又按照空白2针,供试品2针,空白2针,供试品2针的顺序进样,数据如下:[img=只进供试品和空白,690,267]https://ng1.17img.cn/bbsfiles/images/2021/07/202107031139383447_5262_3356999_3.png!w690x267.jpg[/img]令本人百思不得其解,请各位分析解答,谢谢~最后附上检验方法(仪器为安捷伦7890B,色谱柱为安捷伦VF-17ms,砂芯衬管)[img=,690,596]https://ng1.17img.cn/bbsfiles/images/2021/07/202107031143388912_5805_3356999_3.png!w690x596.jpg[/img][img=,690,368]https://ng1.17img.cn/bbsfiles/images/2021/07/202107031143564333_3950_3356999_3.png!w690x368.jpg[/img]
各位老师:乙醇用[url=https://insevent.instrument.com.cn/t/Mp]气相[/url]检测挥发性物质时,进对照品a出来的峰拖尾还分不开是什么原因呢[img]https://ng1.17img.cn/bbsfiles/images/2019/06/201906041012387514_2300_3926493_3.jpeg[/img][img]https://ng1.17img.cn/bbsfiles/images/2019/06/201906041012529407_1811_3926493_3.jpeg[/img][img]https://ng1.17img.cn/bbsfiles/images/2019/06/201906041012539217_9202_3926493_3.jpeg[/img][img]https://ng1.17img.cn/bbsfiles/images/2019/06/201906041012593807_5993_3926493_3.jpeg[/img]
白藜芦醇是多酚类化合物,主要来源于蓼科植物虎杖的根茎提取物。白藜芦醇的测定方法为我司协助客户进行色谱柱筛选所做应用,实验结果表明:INERTSIL ODS-3做白藜芦醇拖尾因子1.1,钻石二代(Diamonsil C18(2))拖尾因子1.028,钻石一代(Diamonsil C18)拖尾因子1.097,极性改性反相色谱柱—思博尔(Spursil C18) 拖尾因子1.076。以下为白藜芦醇检测详细的测试条件和不同色谱柱分析结果,请您参考!样品制备制备方法:对照品溶液的配制:白藜芦醇对照品8mg(精确至0.02mg),准确加入80-90mL甲醇溶解,充分混匀,定容至100mL。 分析条件流动相:水:乙腈=70:30流速:1.0 mL/min柱温:35 ℃检测器:UV 303 nm进样量:10 μL






 留言咨询
留言咨询

 400-860-5168转5114
留言咨询
400-860-5168转5114
留言咨询
 400-860-5168转5114
留言咨询
400-860-5168转5114
留言咨询











 我要推广仪器
我要推广仪器
 下载APP
下载APP