大家使用[url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]时如果用微量进样器进样的话,液体样品进样量是多少?咱们交流一下我们填充柱进样为1微升;毛细柱分流进样0.2或者0.4微升再补充点:不知道各位用的微量进样器是多大量程的,我们用10微升的进样0.2微升总是感觉不太保险
[color=#444444]最近用[url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]检测总是出现这样的问题,同一样品,进样体积一样,但出峰高度差不少,含量变化也很大,一个含量在70%,一个30%左右,能浮动8%左右,这样根本没法定量啊,即使是粗略的定量。为什么重复性会这么差呢,是色谱的问题,还是色谱条件,或者操作的问题?[/color]
各位老师,我最近在苯甲醇氧化反应,今天在检测反应液(主要成分是苯甲醇、苯甲醛、苯甲酸、溶剂为乙酸乙酯)的时候,在其他色谱条件相同的条件下,当进样量为0.2ul的时候,苯甲醇的转化率为89%,但当进样量为0.4ul时,苯甲醇的转化率为79%,当进样量降低为0.1ul的时候,苯甲醇的转化率变的更低,将为60%多,而各进样条件下苯甲醛的选择性基本相同,我想问问各位老师为什么进样量不同时,苯甲醇的转化率会相差这么大,我该以哪一个进样量条件下的转化率为准呢?而一般的进样量是多少?是怎么确定的?我的色谱条件为:进样器:150℃、柱温:280℃、检测器:280℃,手动进样,采用面积归依法计算各组分含量。柱子为SE-54,50m×0.32mm×0.50mm,国产色谱,按理说,同样的样品在相同条件下,含量应该会一样的,怎么我在进样量不同时得出了不同的含量呢?我再问一下,SE-54对苯甲醇、苯甲酸、苯甲醛、乙酸乙酯可以分的开吗?麻烦各位老师指导指导我,谢谢了!
各位老师,大家好,最近在做苯甲醇氧化反应,今天用气相色谱分析苯甲醇、苯甲醛、苯甲酸、乙酸乙酯(用作溶剂)的含量时,其他色谱条件未改变、且都相同的情况下,当进样量为0.2ul时,苯甲醇的含量为89%,但当进样量为0.4ul的时候,苯甲醇的转化率为79%,两次进样量的条件下,苯甲醛的选择性基本相同,我想请教一下各位老师,哪个结果较为准确呢,为啥进样量不同,各组分含量变化会如此之大呢?我的色谱条件为:进样器:150℃、柱温:280℃、检测器(FID):280℃。柱子为毛细管柱SE-54,规格为:50m×0.32mm×0.50um。手动进样,国产色谱,采用面积归依法计算各组分含量的。按理说,同样的样品,含量是定的,怎么进氧量不一样时,结果反而还不一样,请各位老师给我指导指导我该怎么做,造成这种问题的原因是什么?是仪器的问题,还是我方法的问题,我该怎么做才能消除这种问题呢?谢谢了
气象色谱在分析样品时,进样量与“色谱柱的容量”有关吗
[url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]如何选择进样时间和进样量?
[size=18px]在fid检测器里,信号值控制在多少比较合适。我现在进样0.8微升,溶质稀释4倍,而溶剂峰峰高约6万,担心进样量过大没有增大进样量,比较难以进行微量分析。[/size][size=18px]是否有句话说[url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]填充柱可接受的单个组分的量是1μg,那这个组分是否包括溶剂呢?[/size]
[color=#444444]我最近在苯甲醇氧化反应,今天在检测反应液(主要成分是苯甲醇、苯甲醛、苯甲酸、溶剂为乙酸乙酯)的时候,在其他色谱条件相同的条件下,当进样量为0.2ul的时候,苯甲醇的转化率为89%,但当进样量为0.4ul时,苯甲醇的转化率为79%,当进样量降低为0.1ul的时候,苯甲醇的转化率变的更低,将为60%多,而各进样条件下苯甲醛的选择性基本相同,我想问问各位老师为什么进样量不同时,苯甲醇的转化率会相差这么大,我该以哪一个进样量条件下的转化率为准呢?而一般的进样量是多少?是怎么确定的?我的色谱条件为:柱温:150℃、气化温度:280℃、检测器:280℃,手动进样,采用面积归依法计算各组分含量。柱子为SE-54,50m×0.32mm×0.50mm,国产色谱,[/color][color=#444444]按理说,同样的样品在相同条件下,含量应该会一样的,怎么我在进样量不同时得出了不同的含量呢?我再问一下,SE-54对苯甲醇、苯甲酸、苯甲醛、乙酸乙酯可以分的开吗?麻烦各位老师指导指导我,谢谢了![/color][color=#444444]我问问各位老师,我该怎么做才能避免这种问题的发生,还有,我问一下造成这种问题的原因是什么,是仪器的问题呢?还是我的操作问题呢?谢谢了[/color]
[url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]的进样量一般选多少,液体我一般2微升,气体80微升,对吗,期待高手赐教
[url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]进样量或进样方式不一样会影响某种物质的保留时间吗?
我想用安捷伦的气相色谱分析气相产品,主要是水蒸气和乙烯,丙烯,苯乙烯的混合物,可能还有会其他的低碳烃。水蒸气可以先冷凝下来,不用考虑。分析这些物质,阀动进样一次需要多少样品,如果用气密性的进样针手动进样又需要多少样品?安捷伦提供的定量环和手动进样针的范围非常大,不知道在什么范围内比较合适。因为搭建反应体系很麻烦,我想尽量建一个小反应体系,能把产物定量分析出来就可以。谢谢各位
气相色谱在不超载的情况下,进样量为了防止太多或太少,有没有个大致标准?有人说是满量程的80% 对不?
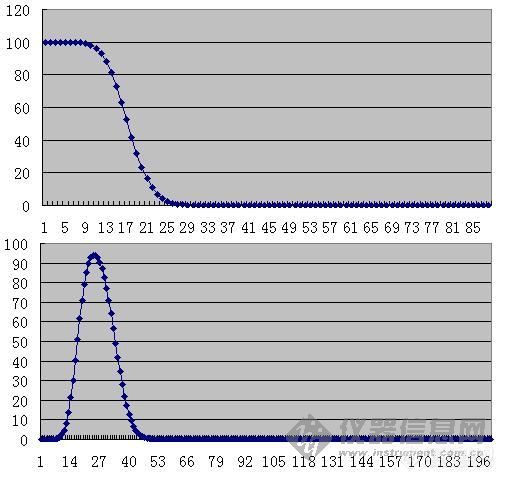
其他讲座资料看[url=http://www.instrument.com.cn/bbs/detail.asp/threadid/1679222/forumid/25/year/2009/query/search] 学习[url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]跟yuen72老师入门[/url]在塔板理论脉冲进气的情况下,样品进样都在第0块塔板上。如此,第0块塔板体积应该等于进样体积。但是,在毛细管柱的进样上,明显并不如此。以0.32mm色谱柱正常柱效为3300块塔板每米来看,每块塔板高度应为0.3mm。如果考虑进样量为0.2ml,分流比为50倍,则进入色谱柱的样品体积是为0.004ml,如此应该占有50mm的色谱柱,按照塔板理论假设,理论塔板高度应为50mm。为什么会出现这样的差异呢?显然问题来自与理论假设中的脉动进样。通常,我们把实际进样体积占色谱柱高度与理论塔板高度之比称为进样宽度,这里即50mm/0.3mm=167块塔板。在样品逐渐进入色谱柱的时候,溶解和解析的过程是不断进行的,实际的样品进样过程中,已经形成了色谱峰的形状。如下图,上面的是进样宽度为100块塔板高度情况下,进样完毕时组分浓度的柱内分布。当包括进样在内共有200块塔板体积载气进入色谱柱后,形成的色谱峰成为下面的形状。注意,图中横坐标为色谱柱头0至n块塔板。[img]http://ng1.17img.cn/bbsfiles/images/2009/03/200903060121_136950_1641581_3.jpg[/img]理论塔板高度事实上是与进样无关的,只与载气流速、纵向扩散速度和传质速度有关,即速率理论:H = B/u + Cu 。因此在相同线速度(纵向扩散相同)和相比(液膜厚度与内径成正比)的情况下,存在理论塔板高度与毛细管色谱柱内径成正比的关系,因为此时传质距离正好与内径成正比。[B]因此,厚液膜的色谱柱,因为增加了液相厚度而增加了液相传质阻力,因此并不能提供更好的理论塔板数。相反的,厚液膜色谱柱理论塔板数(即柱效率)要略低一些。[/B]这与我们实际工作似乎略有出入,事实上,实验室中经常发现厚液膜的柱子具有更好的分离度R。但这与理论并不冲突,发生这样情形的原因在于进样量。
[color=#444444][url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]对进样量浓度有要求吗?我想先用[url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]分析下,看看上面的峰是否分离的较好。所以,请问下刚开始进样样品稀释到多少倍较好,一般都是按多少的比例?(我这个样品中有高沸点组分达350度)才能使得图中主要的峰看起来更突出。谢谢[/color]
1、同样分子量的样品,支化度大的分子和线形分子哪个先流出色谱柱? 2、讨论进样量,色谱柱的流速对实验结果有无影响?3、温度,溶剂的优劣对高聚物色谱图的位置有什么影响4、GPC的分离机理与气相谱的分离机理有什么不同
毛细管色谱柱: CP-WAX58(FFAP)50m*0.25mm*0.2µ m的最高进样量是多少?载气流速:1.1ml/min,分流比:1:40
[color=#444444]HPLC用自动进样器进样,设定进样量是5ul,样品共有100ul(在普通色谱瓶里加了微量小玻璃管),但进一次样就少了50ul。不知道是否哪里出问题了还是自动进样器本来就是要多吸取样品的?以前都用1ml的样品量,进样多少没有注意。[/color]
单位想采购一些色谱仪检定用的微量进样器,请问那个牌子的微量进样器质量比较好,或者那个厂家的比较好,请熟悉的朋友告知一下,我们平头和尖头都需要的,最好有销售的联系电话,谢谢。
本人做医药农药中间体研发实验自己做的[url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]进样一针获得的产品含量和中控分析中心人员的获得数据不同,进样量一样,但是分析人员的数据总是比我的值高,请问哪位朋友能给解释一下,是不是柱子有问题?
今天我们的色谱仪器进行鉴定,鉴定人员说进样量很大的情况下会把色谱柱弄断,不知这种说法有没有道理?我还是第一次听说,大家有没有听说过呢
进样口与色谱柱具体怎么搭配。可以在电脑操作系统中设置色谱中一个进样口接两个色谱柱吗?
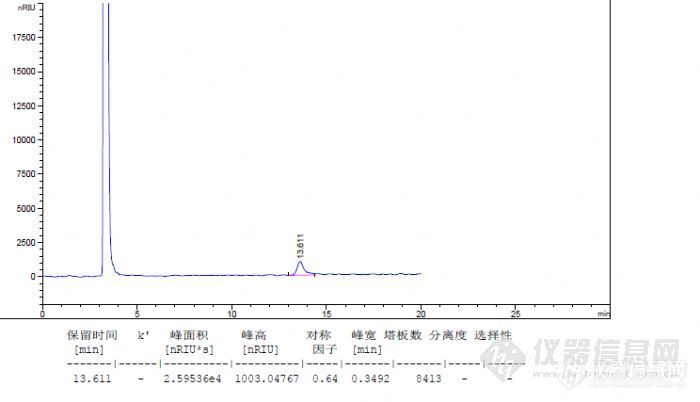
前言:按照10版药典方法检测乳糖的含量,按照系统适应性下的要求,乳糖与蔗糖达到分离要求,乳糖的理论踏板数不小于5000。下面主要考察乳糖在测试过程中,相同样品浓度下不同的进样体积对柱效的影响和不同浓度下相同进样体积对柱效的影响。测定方法:色谱柱:Ultimate®XB-NH2-3,4.6×250mm,5μm; 温度:柱温:45度,示差检测器:40度;流动相:乙腈-水=70:30; 流速:1.0mL/min; 进样量:10μL。样品配制:系统适用性:精密称取乳糖0.0255g和蔗糖0.0254g置于25ml容量瓶中加水溶解并稀释至刻度摇匀,过滤,即得。含量测定样品溶液:精密称取样品粉末0.0252g于25ml容量瓶中,用水溶解并定容,过滤即得;含量测定对照品溶液:精密称取对照品粉末0.0254g于25ml容量瓶中,用水定容,过滤即得按照测定要求,按照测定要求样品浓度为1mg/ml,进样体积:10ul考虑到我的仪器的定量环是20ul,因为是手动进样,进样量10ul可能在计算含量的时候峰面积不稳定计算会产生误差。考虑把样品浓度配制成0.5mg/ml,调整进样体积为20ul;下面上一张系统适用性图:http://ng1.17img.cn/bbsfiles/images/2012/11/201211032333_401206_1938106_3.jpg重复了好几针,分离度和保留时间没改变,柱效依然达不到5000.乳糖对照品0.5mg/ml 进样量20ulhttp://ng1.17img.cn/bbsfiles/images/2012/11/201211032338_401211_1938106_3.jpg测试乳糖对照品,乳糖的柱效也是比较低的。考虑到示差检测器是比较难稳定的,也考虑氨基柱在平衡中是比较难平衡的,所以采用过夜循环平衡方法进行测定。测试乳糖对照品0.5mg/ml 进样量20ulhttp://ng1.17img.cn/bbsfiles/images/2012/11/201211040004_401226_1938106_3.jpg平衡了一夜但的柱效一点都没有改善。柱效依然不能满足要求。这个过程考虑到是否是氨基柱的问题,排除了氨基柱在使用过程中的氨基脱落导致柱效的原因,为了排除色谱柱的原因换新柱子测试,柱效还是不能满足要求。百思不得其解中,想到样品质量对柱效影响,还是进样体积对柱效是否有影响。为了证明是相同质量下,不同进量是否对柱效存在影响。下面配制1mg/ml的浓度分别进样20ul和10ul的体积。乳糖对照品1.0mg/ml 进样量20ul http://ng1.17img.cn/bbsfiles/images/2012/11/201211041842_401303_1938106_3.jpg乳糖对照品1.0mg/ml 进样量10ulhttp://ng1.17img.cn/bbsfiles/images/2012/11/201211041848_401305_1938106_3.jpg进样体积减少10ul发现柱效提高一倍多。结论:从以上实验结果分析比较当乳糖含量样品体积为20ul,样品浓度1mg/ml,柱效:3982;0.5mg/ml,浓度,柱效4253;当乳糖浓度1mg/ml的浓度进样为20ul柱效:3982;进样量10ul柱效:8413,进样体积减小一半,柱效提高一倍。说明在夜想色谱柱具有一定的饱和容量下,最大进样体积,也要决定于化合物在固定相和流动相中平衡,通过上面实验证明,随着进样量的增大,柱效降低。当进样量超出色谱柱饱和容量时,柱效也会降低。对于一定的样品量,进样体积增大,柱效降低。当进样量超出色谱柱上样最大进样体积时,柱效会有明显降低,影响分离结果。当然对于分析色谱而言,所需要的进样量和进样体积都是很小的,所以在平时很少观察到这点现象。可以看出进样量和进样体积对定性定量中对色谱柱柱效的影响。发现晚上传图太慢了, 所以今天晚上才把图传上来,供各位色友分享。
液相色谱一般都有定量环 20ul的居多 但是我们进样有时候进20有时候进10的 进20ul时候、大家一般用20ul定量环时候进多少 有人说 进三倍量 那如果近10ul的是不还要换定量环啊 我一直都是20的 从没有换过 一般也没有按照进3倍量!
我们用的是安捷伦1100的高效液相色谱仪,在做一个软膏剂的时候,标准规定的浓度进样10μl,峰形有前延峰,把浓度稀释一半,进样10μl,峰的对称性在1.0左右,如果再把浓度稀释一半,进样20μl的话,峰形又不对称了。大家分析一下是什么原因?液相色谱的进样量与峰形之间的关系怎样?(药品名字是“复方地塞米松乳膏”,内标法测定地塞米松含量,内标物是甲睾酮)
当用[url=https://insevent.instrument.com.cn/t/5p][color=#3333ff]液相色谱[/color][/url]溶剂极性强度大的试剂,如纯甲醇、纯乙腈,纯乙醇,而分析体系中以水为主时,如果样品进样量大,如定量管为20ul,此时单一的纯物质会出现双峰,第二峰比第一峰小(每次都不太一样),且拖尾,保留时间会提前(相对进样量少而言),将进样量减少一半以上,峰型将变为正常。这是样品的溶剂与流动相极性相差太大,而流动相来不及将其稀释达到平衡造成的,此情况下需要减少进样量。另一个原因是,进样量不一定大,但绝对量很大,色谱图上的双峰紧靠在一起,基本上齐高,不拖尾(如果出峰很快,也可能是色谱柱问题)。将样品稀释再进样就可以了,这是由于进样量过大,色谱柱过载造成的。
我使用的[url=https://insevent.instrument.com.cn/t/3p][color=#3333ff]离子色谱仪[/color][/url]是手动进样的,戴爱ICS1500,想请问各位高手,一般进样量是多少啊,进样量对出来的峰有影响吗?仪器重现性很不好,怎么同一个样品,第一次进样检出来浓度很高,第二次进样又检不出了,请问事怎么回事啊?
本人刚接触液相色谱,想要检测某饮料中某一物质的含量采用外标法,该物质的大体含量不清楚,看了几篇参考文献说外标法中浓度范围最好包括样品浓度,这该怎么做呢?难道扩大外标法中的浓度范围吗,这样好像比较麻烦又不见得准确啊?还有就是进样量应该怎么确定啊?
请问怎样消除[url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]毛细管柱因液体进样量太大(10μl)而导致的目标物在仪器上的残留污染,谢谢![em0810]
[color=#444444][url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]手动进样,连续进了6针含量都不一样,误差在5左右,而且该很低的含量反而多。今天色谱基线还出现了负峰,请问是什么原因,该如何解决?求大神解答。[/color]
高效液相色谱进样量一般为多少啊







 我要推广仪器
我要推广仪器
 下载APP
下载APP