请问,安捷伦的固相微萃取纤维和色谱科的通用吗?谢谢!
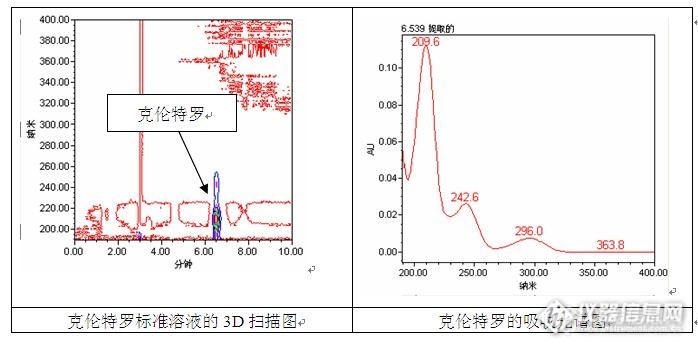
[align=center]固相萃取-高效液相色谱法测定蜂蜜中克伦特罗残留量及条件优化[/align][align=left]1、前言 克伦特罗(Clenbuterol)既不是兽药,也不属于添加剂,而是一种激素类物质,俗称“瘦肉精”,该物质能促进动物体内脂肪分解代谢,增加蛋白质合成,提高瘦肉率,然而人体食用高残留量的内脏组织或者累计摄入量超过一定值时便可能引发食物中毒事件。1997 年我国明令禁止在畜牧行业生产、销售和使用克伦特罗,但是非法使用克伦特罗的事件仍时有发生。 目前,现行有效的标准中测定克伦特罗的方法有酶联免疫法、胶体金免疫层析法、液相色谱法和质谱法,针对不同的样品基质和实验室条件可以选择合适的测定方法。国家标准GB/T 22944-2008中采用液相色谱-串联质谱法测定蜂蜜中克伦特罗的残留量,但是实验室没有[url=https://insevent.instrument.com.cn/t/Yp][color=#3333ff]液质[/color][/url],关于高效液相色谱法测定蜂蜜中克伦特罗的文献也不多,于是尝试建立固相萃取-高效液相色谱法测定蜂蜜中克伦特罗的方法并对测试条件进行优化。2、实验方法2.1 仪器和试剂 Waters e2695液相色谱仪(主要包括2998光电二极管矩阵检测器,柱温箱,自动进样器,自动脱气四元梯度泵等);微型漩涡混合仪;12位固相萃取真空装置;12位干浴氮吹仪; Millpore超纯水系统。 甲醇中盐酸克伦特罗标准溶液(250μg/mL,坛墨质检);甲醇、乙酸乙酯和乙酸为色谱纯;乙酸钠、乙酸铵、磷酸二氢钠、氨水和磷酸为分析纯;实验用水为Millpore超纯水系统制得,18MΩ• cm,25 ℃。2.2色谱条件 色谱柱:CNW Athena C18-WP(250mm×4.6mm ,5μm) 流动相:甲醇:磷酸二氢钠(0.02mol/L)=40:60 流速:1.0mL/min 进样体积:20μL 柱温:30℃ 检测波长:210nm3 结果与讨论3.1 检测波长的确定 采用Waters 2998二级管阵列检测器的3D扫描功能,在波长190~400nm范围内对高浓度的克伦特罗标准工作溶液进行测定,并在克伦特罗出峰位置提取光谱图,见下图,从图中可以看出,克伦特罗的最大吸收波长在210nm附近,因此选择210nm作为检测波长,此时克伦特罗的响应值最大。[/align][align=center][img=,690,345]http://ng1.17img.cn/bbsfiles/images/2017/09/201709301354_01_1669358_3.jpg[/img][/align][align=left]3.2 流动相的选择3.2.1甲醇-水为流动相3.2.1.1 流动相pH值对测定结果的影响 首先参考GB/T 5009.192-2003第二法中的色谱条件,以甲醇-水为流动相进行测试,然而结果并不理想,克伦特罗标准溶液在此流动相下并未出峰,几番尝试后决定更换流动相。看到几个采用质谱测定的标准方法均在流动相中加入了酸,于是仍然以甲醇-水为流动相,往水中加入不同体积的磷酸溶液,考察pH值对克伦特罗测定结果的影响(甲醇:水=30:70),测定结果见下图。[/align][align=center][img=,583,845]http://ng1.17img.cn/bbsfiles/images/2017/09/201709301356_01_1669358_3.jpg[/img][/align][align=left] 从上述测定结果中可以发现,未调pH时色谱图中并没有明显的色谱峰,流动相中加入磷酸后克伦特罗在8min左右出峰;随着磷酸加入量的增加,其出峰时间提前并且峰高与峰面积也随着增加;当进一步提高流动相中磷酸含量时,克伦特罗的出峰时间逐渐延长,但是峰面积保持不变。这可能是由于克伦特罗呈弱酸性,在水溶液中以离子形态存在,降低了在C18色谱柱上的保留行为,而磷酸的加入能够抑制克伦特罗的电离,使其以分子的形态存在,增加克伦特罗在C18色谱柱上的保留,并改善峰形。当pH=3.5时,克伦特罗呈部分解离的状态,因此虽然能够出峰,但是峰面积偏小,而当pH=3.0时,克伦特罗则全部以分子形态存在,峰面积保持不变。随着磷酸加入量的增加,克伦特罗的出峰时间延长,峰展宽变大,峰高变小,方法的灵敏度也随着降低。3.2.1.2 流动相比例对测定结果的影响 流动相中有机相比例对高效液相色谱的分离行为有很大影响,调节流动相比例可以改善待测组分的峰形、出峰时间以及与杂质组分的分离度,因此实验中考察了有机相比例对克伦特罗测定结果的影响(pH=2.8),测定结果见下图。从图中可以看出,提高流动相中甲醇含量对克伦特罗的出峰时间有很大的影响,当流动相中甲醇含量为20%时,克伦特罗的出峰时间为17min左右,而当流动相中甲醇含量为45%时,克伦特罗的出峰时间提前到3min左右,而且与溶剂峰重叠,不利于定性和定量分析。[/align][align=center][img=,578,826]http://ng1.17img.cn/bbsfiles/images/2017/09/201709301359_02_1669358_3.jpg[/img][/align][align=left]3.2.1.3 方法重复性测试 根据上述测试结果,初步决定采用甲醇:水(pH=2.8)=30:70的流动相条件进行测试,然而在测试过程中发现,此流动相条件下克伦特罗的保留时间不稳定,随着进样次数的增加,保留时间不断前移,6针进样后克伦特罗保留时间的相对标准偏差(RSD)值为1.3%。产生保留时间漂移的原因可能有两种(1)色谱柱的性能下降,流动相中加了磷酸,色谱柱平衡所需的时间比较长(这是一根服役了很久的色谱柱);(2)此流动相条件的缓冲能力弱,在线混合以及样品的加入导致流动相的pH值发生变化,从而保留时间不稳定。于是修改流动相条件,pH值保持不变,将流动相中甲醇的比例由30%提高至40%,重新考察方法的重复性,实验结果见下图。实验表明,甲醇:水(pH=2.8)=40:60时方法具有较好的重复性,连续6针进样,克伦特罗保留时间的RSD值为0.1%。[/align][align=center][img=,589,419]http://ng1.17img.cn/bbsfiles/images/2017/09/201709301401_01_1669358_3.jpg[/img][/align][align=left]3.2.2甲醇-磷酸盐为流动相 通过上述实验,初步确立了以甲醇-水为流动相测定克伦特罗的高效液相色谱条件,并对部分实验参数进行了优化,然而上述实验结果均是针对克伦特罗标准工作溶液进行的测定,样品成分单一,没有杂质干扰,因此不用考虑杂质与克伦特罗之间的分离度。色谱条件为甲醇:水(pH=2.8)=40:60时虽然解决了方法重复性的问题,但是从色谱图中可以看到,此时克伦特罗的出峰时间较早,仅需3.5min左右就能出峰,而且出峰时间早于溶剂峰,在实际样品分析中很容易受到杂质峰的影响,此方法是否适合测定蜂蜜中的克伦特罗残留量还需进一步验证。 在对蜂蜜样品测定验证之前,先尝试寻找是否有更合适的流动相。以甲醇-水为流动相进行测试时,磷酸的加入对克伦特罗的出峰影响较大,于是尝试改用甲醇-磷酸盐为流动相进行下一步测试。从流动相pH值、流动相比例和方法重复性3个方面对方法进行考察,磷酸盐选用0.02mol/L的磷酸二氢钠,通过磷酸调节流动相的pH值,实验结果见下图。实验表明,以甲醇-磷酸二氢钠(0.02mol/L)为流动相时,有机相比例对克伦特罗出峰时间也有很大的影响,pH值的影响较小,因此后续的实验中直接采用甲醇-磷酸二氢钠(0.02mol/L)为流动相,未对流动相的pH值进行调节。当甲醇:磷酸二氢钠(0.02mol/L)=40:60时,连续6针进样,克伦特罗保留时间的RSD值为0.1%,测试方法具有较好的重复性,同时,克伦特罗的出峰时间远离溶剂峰,可以减小样品中杂质组分对待测物质测定的干扰。[/align][align=center][img=,597,550]http://ng1.17img.cn/bbsfiles/images/2017/09/201709301403_01_1669358_3.jpg[/img][/align][align=left]3.2.3甲醇-乙酸盐为流动相 在查阅文献的过程中发现,也有老师采用乙酸盐缓冲溶液对克伦特罗进行测定,于是决定试一下效果。同样,从流动相pH值、流动相比例和方法重复性3个方面对方法进行考察,乙酸盐选用0.02mol/L的乙酸铵,通过乙酸调节流动相的pH值,实验结果见下图。实验表明,采用甲醇-乙酸铵(0.02mol/L)为流动相时,有机相比例对克伦特罗出峰时间有很大的影响,pH值的影响较小,方法的重复性较好,但是此时基线噪音较大,峰高变小,影响方法的检出限。[/align][align=center][img=,581,588]http://ng1.17img.cn/bbsfiles/images/2017/09/201709301405_01_1669358_3.jpg[/img][/align][align=left] 通过上述实验,分别以甲醇-水、甲醇-磷酸盐和甲醇-乙酸盐为流动相,建立了高效液相色谱法测定克伦特罗的方法,并对部分实验条件进行了优化,在实验中发现了各自方法的优缺点,哪种方法更适合蜂蜜中克伦特罗的测定,还需用蜂蜜样品进行验证。3.3 样品前处理 蜂蜜中通常不含有克伦特罗,即使有,其残留值也很小,而且蜂蜜为半固态粘稠样品,无法直接进样测定,因此需要对蜂蜜样品进行前处理。前处理过程主要包含提取、富集、净化和浓缩4个步骤,此次实验蜂蜜中克伦特罗的提取方法参考GB/T 22944-2008:称取约2g蜂蜜样品于50mL离心管中,加入20mL乙酸钠缓冲溶液(0.2mol/L,pH=5.0),漩涡混匀,蜂蜜完全溶解后备用。 样品中克伦特罗残留量富集、净化的方法有很多种,本次实验分别参照标准GB/T5009.192-2003、SN/T1924-2007、SN/T1924-2011以及GB/T22944-2008,采用CNWBONDWCX(500mg,6mL)、CNWBOND SCX(500mg,6mL)、CNW Poly-Sery MCX(60mg,3mL)和CNW Poly-SeryHLB(500mg,6mL)固相萃取柱对蜂蜜中的克伦特罗残留量进行富集、净化,其中SN/T1924-2007标准已作废,但是标准中所涉及的富集净化方法也曾出现在上海安谱实验科技股份有限公司(以下简称上海安谱)的技术应用文章中,因此对此方法也做了尝试。同时,采用Oasis MCX(60mg,3mL)和OasisHLB(200mg,6mL)固相萃取柱对蜂蜜中的克伦特罗残留量进行富集、净化,简单比较了不同品牌固相萃取柱在净化效果、回收率等性能方面的差异。[/align][align=center][img=,578,358]http://ng1.17img.cn/bbsfiles/images/2017/09/201709301407_01_1669358_3.jpg[/img][/align][align=left] 固相萃取柱所用的填料不同,活化和净化方法也有所区别,具体实验方法如下: (1)WCX固相萃取柱 依次用5mL乙醇和5mL水活化固相萃取柱,将上述蜂蜜提取液全部转移至活化后的固相萃取柱中,分别用5mL水和5mL 乙醇淋洗固相萃取柱,弃去淋洗液,真空抽干2min,最后用10mL氨水-乙醇溶液(体积比2:98)进行洗脱,收集洗脱液。洗脱液在50℃下氮气吹干,准确加入1mL水溶解残渣,过0.22μm滤膜后,进样测定。 (2)SCX固相萃取柱 依次用5mL甲醇、5mL水和5mL0.03mol/L盐酸溶液活化固相萃取柱,将上述蜂蜜提取液全部转移至活化后的固相萃取柱中,分别用5mL水和5mL甲醇淋洗固相萃取柱,弃去淋洗液,真空抽干2min,最后用10mL氨水-甲醇溶液(体积比5:95)进行洗脱,收集洗脱液。洗脱液在50℃下氮气吹干,准确加入1mL水溶解残渣,过0.22μm滤膜后,进样测定。 (3)MCX固相萃取柱 依次用3mL甲醇、3mL水和3mL 0.1mol/L盐酸溶液活化固相萃取柱,将上述蜂蜜提取液全部转移至活化后的固相萃取柱中,分别用3mL 0.1mol/L盐酸溶液、3mL水和3mL50%甲醇淋洗固相萃取柱,弃去淋洗液,真空抽干2min,最后用5mL氨水-甲醇-乙酸乙酯溶液(体积比5:45:50)进行洗脱,收集洗脱液。洗脱液在50℃下氮气吹干,准确加入1mL水溶解残渣,过0.22μm滤膜后,进样测定。 (4)HLB固相萃取柱 依次用5mL甲醇和5mL水活化固相萃取柱,将上述蜂蜜提取液全部转移至活化后的固相萃取柱中,分别用5mL水和5mL甲醇淋洗固相萃取柱,弃去淋洗液,真空抽干2min,最后用10mL氨水-甲醇溶液(体积比5:95)进行洗脱,收集洗脱液。洗脱液在50℃下氮气吹干,准确加入1mL流动相溶解残渣,过0.22μm滤膜后,进样测定。[/align][align=center][img=,578,190]http://ng1.17img.cn/bbsfiles/images/2017/09/201709301408_01_1669358_3.jpg[/img][/align][align=left]3.4 蜂蜜样品的测定3.4.1甲醇-水为流动相 以甲醇-水(pH=2.8)为流动相,测定固相萃取柱净化后的蜂蜜及蜂蜜加标样品,考察该色谱条件对蜂蜜中克伦特罗残留量测定的影响。 当甲醇:水(pH=2.8)=40:60时,测定结果见下图。从图中可以看出,蜂蜜样品提取后直接进样测定色谱图中,在克伦特罗保留时间处有一个很大的杂质峰,虽然该杂质峰经HLB固相萃取柱净化后消失,但是实际检测时该杂质峰对克伦特罗仍然存在隐患,如果净化不干净,就可能出现假阳性的结果,同时,在该流动相条件下,样品在2~6min内有许多杂质峰,影响克伦特罗与杂质组分之间的分离度。对比HLB与MCX固相萃取柱净化后测定的色谱图,在此色谱条件下,HLB的净化效果要优于MCX固相萃取柱,蜂蜜样品经HLB固相萃取柱净化后杂质峰明显减少,MCX固相萃取柱净化后仍有杂质组分会对克伦特罗的测定产生干扰。[/align][align=center][img=,581,863]http://ng1.17img.cn/bbsfiles/images/2017/09/201709301411_01_1669358_3.jpg[/img][/align][align=left] 改变流动相比例,测定蜂蜜加标样品经MCX固相萃取柱净化后的样品,改善克伦特罗与杂质间的分离度,实验结果见下图。从图中可以看出,当甲醇:水(pH2.8)=30:70时克伦特罗与杂质组分的分离度较差,不能实现基线分离;当甲醇:水(pH2.8)=20:80时,克伦特罗与杂质组分虽然能实现基线分离,而且附近没有杂峰干扰,但是此时克伦特罗的峰高变小,灵敏度变低,不利于低浓度克伦特罗残留量的检查。[/align][align=center][img=,584,295]http://ng1.17img.cn/bbsfiles/images/2017/09/201709301413_02_1669358_3.jpg[/img][/align][align=left]3.4.2甲醇-乙酸铵为流动相 以甲醇-乙酸铵(0.02mol/L)为流动相,测定MCX固相萃取柱净化后的蜂蜜加标样品,考察该色谱条件对蜂蜜中克伦特罗残留量测定的影响,实验结果见下图。从图中可以看出,当流动相为甲醇:乙酸铵(0.02mol/L)=60:40时,克伦特罗的测定受到杂质组分的影响很大,克伦特罗出峰时基线较高,而且与杂质组分不能基线分离;当流动相为甲醇:乙酸铵(0.02mol/L)=40:60时,样品中克伦特罗的灵敏度明显降低。[/align][align=center][img=,586,354]http://ng1.17img.cn/bbsfiles/images/2017/09/201709301414_01_1669358_3.jpg[/img][/align][align=left]3.4.3甲醇-磷酸二氢钠为流动相 以甲醇-磷酸二氢钠(0.02mol/L)为流动相,当甲醇:磷酸二氢钠(0.02mol/L)=40:60时,测定固相萃取柱净化后的蜂蜜及蜂蜜加标样品,考察该色谱条件对蜂蜜中克伦特罗残留量测定的影响,实验结果见下图。从图中可以看出,当甲醇:磷酸二氢钠(0.02mol/L)=40:60时,克伦特罗与样品中的杂质实现基线分离,其保留时间附近的干扰组分较少,灵敏度能够满足残留量检测的要求。同时,在此色谱条件下,蜂蜜加标样品只需经过简单的提取便能直接测定,给高残留值蜂蜜样品的测定提供了便捷的方法(直接提取进样时的加标量远高于采用固相萃取柱净化时的加标量)。结合上述实验,最终本实验选择采用甲醇-磷酸二氢钠(0.02mol/L)为流动相对蜂蜜中克伦特罗残留量进行测定。[/align][align=center][img=,578,594]http://ng1.17img.cn/bbsfiles/images/2017/09/201709301416_01_1669358_3.jpg[/img][/align][align=center][img=,580,634]http://ng1.17img.cn/bbsfiles/images/2017/09/201709301417_01_1669358_3.jpg[/img][/align][align=left] 找到合适的流动相后,在该色谱条件下对固相萃取柱的净化效果进行考察。从上图可以看出,乙酸钠缓冲溶液提取后直接进样的色谱图中虽然也能检出克伦特罗,但是其加标量较大(约为固相萃取柱方法的20倍,该实验主要是为了考察杂质对待测物质的干扰),实际样品中克伦特罗的残留值远小于此次的加标量,因此实际样品测定时需要采用固相萃取柱对克伦特罗残留量进行富集、净化和浓缩。对比不同填料固相萃取柱对蜂蜜加标样品净化后测定的色谱图可以发现,以甲醇-磷酸二氢钠(0.02mol/L)为流动相时,CNWBOND SCX固相萃取柱净化后杂质组分的响应值依然很大,SN/T 1924-2007中采用了C18和SCX固相萃取柱串联的方法进行净化,而本实验仅采用了SCX固相萃取柱进行净化,这可能是导致杂质去除不完全的原因之一;采用其他几种固相萃取柱净化后,色谱图中杂质峰的响应值明显降低,其中采用WCX固相萃取柱净化后的色谱图中杂质少,基线比较平整。对比相同填料不同品牌固相萃取柱净化后的色谱图可以发现,两种品牌的固相萃取柱对杂质去除能力难分伯仲。3.5 标准曲线的绘制 准确吸取1.0mL盐酸克伦特罗标准溶液于25mL容量瓶中,用水稀释成浓度为10.0μg/mL的标准储备溶液。吸取适量体积克伦特罗标准储备溶液,用水稀释配成相应浓度的标准工作溶液,并按上述选定的色谱条件进行测定。以样品峰面积Y(mV)对质量浓度X(μg/mL)作图,得到线性回归方程Y=146893X+311,相关系数R2=0.9991,结果表明:克伦特罗含量在0.10~1.00μg/mL之间时,该方法呈现良好的线性关系。标准曲线见下图。[/align][align=center][img=,581,408]http://ng1.17img.cn/bbsfiles/images/2017/09/201709301418_01_1669358_3.jpg[/img][/align][align=left]3.6 回收率的测定 称取约2g蜂蜜样品,共12份,往每份样品中加入0.50mL浓度为1.0μg/mL的克伦特罗标准工作溶液,样品经乙酸钠缓冲溶液提取后,分别采用上述6种固相萃取柱对样品进行净化,每种固相萃取柱做2平行,并以甲醇-磷酸二氢钠(0.02mol/L)为流动相进行测定,考察固相萃取柱的回收率,实验结果见下表。测定结果表明,6种固相萃取柱均具有较好的回收率,回收率大于85%,其中MCX和HLB固相萃取柱的回收率明显高于其他两种填料的固相萃取柱;实验中所使用的相同填料不同品牌的固相萃取柱回收率结果相近。[/align][align=center][img=,690,126]http://ng1.17img.cn/bbsfiles/images/2017/09/201709301420_02_1669358_3.jpg[/img][/align][align=left]4、小结 (1)本实验建立了固相萃取-高效液相色谱法测定蜂蜜中克伦特罗的方法,分别以甲醇-水、甲醇-乙酸盐和甲醇-磷酸盐为流动相,考察了流动相对测定结果的影响。实验结果表明,以甲醇-水为流动相时,流动相对pH值的缓冲能力较弱,克伦特罗的保留时间发生漂移;以甲醇-乙酸盐为流动相时,基线噪音较大,方法灵敏度低;以甲醇-磷酸盐为流动相,方法重复性好,灵敏度较高。 (2)实验中参考不同的标准方法对蜂蜜样品进行前处理,考察了前处理方法对样品净化和回收率影响,同时比较了固相萃取柱性能和品牌对蜂蜜中克伦特罗残留量测定的影响。实验结果表明,固相萃取柱性能对蜂蜜中克伦特罗残留量的测定结果有很大的影响,4种不同填料的固相萃取柱中,MCX和HLB固相萃取柱的回收率较高,WCX和SCX固相萃取柱的回收率略低。 (3)实验中考察了WCX和SCX固相萃取柱的回收率较低的原因:WCX固相萃取柱在乙醇淋洗的过程中会有部分克伦特罗被淋洗下来,从而回收率降低;采用SCX固相萃取柱净化时,洗脱液的碱性需要足够强,否则洗脱不完全,但是提高洗脱液的碱性后,杂质也会一同被洗脱下来。 (4)通过此次实验可以发现,流动相条件和固相萃取柱的选择对样品测定具有很大的影响,不同的流动相其洗脱能力不一样,截止波长也会有差别,实验中以甲醇-乙酸铵为流动相时基线噪音明显大于其中两种流动相;同样是MCX净化后的样品,以甲醇-磷酸盐为流动相时测定得到的样品色谱图中杂质要少于其他两种流动相。 (5)液相色谱虽然对克伦特罗具有较高的响应,但是与质谱相比,质谱具有更高的响应,因此对于残留量很低的样品,还是需要用质谱进行验证,液相方法可以作为前期的筛查手段,[/align]
1.1 正相色谱 八十年代初,人们使用的正相色谱固定相硅胶和吡啶硫氰酸镍盐的络合物晶体能与芳香族化合物形成包合物用于分离芳香族含氮异构体和胆甾醇晶体。已有人[1]将焦炭吸附剂作为填料和键合硅胶作过比较并研究其热力学机理。具有离子化或非离子化功能团的大孔聚合物也开始应用于液相色谱,这些聚合物在整个酸碱范围内稳定,其中一个缺点是当溶剂改变时,它们会热胀冷缩,但是其主要的分析方面用途,即从水中收集痕量研究化合物是没有问题的,通过适当的处理装入色谱柱,一些物质出现过度的峰带变宽,而另一些则出现尖、窄的峰带和高分离度,在强碱性大孔树脂中120分钟内可分离100 种尿液中紫外吸收物质[2]。有人将各种链长度的碳氢配位体键合到硅胶上,根据链长短效应研究保留值的影响,也有过类似的报道,将C22 键合相以及制备键合相担体各种条件作过比较,其键长度和吸附自由能成线性关系。一般地,碱溶液会破坏硅胶,烷基胺比季铵盐更会腐蚀硅胶填料,故通过加填有5mm 硅胶的短预柱来洗脱碱性溶液。Kataev等[3]用聚三氟苯乙烯涂成的硅胶微粒基质并应用到卤代芳烃、多肽和蛋白质的分离。1993年11月,Majors 讨论了在日本发展迅速的HPLC 聚合物填料。Hosoya 制备了大孔聚合乙烯—对—叔丁基苯甲酸丁酯微球和两种别的聚合物微球的性能以及更多的C18HPLC固定相。1.2 反相色谱 固定相的研究进入九十年代更是热火朝天。这方面的研究报道远远超过流动相的研究,人们不断探索保留过程及相关烷基键结构的复杂性和可能制得的各种新型固定相。Bolok 和其合作者从统计技术研究反相色谱柱的老化过程。Montes 等评价了一种硅氢化物介质的硅氢烯制备烷基键合相,更早时候基质制备可供参考。Moriyama等评价了用2mm硅胶制得的TSK胶Super ODS新型反相色谱柱的特性,他们论述了如何分离手性化合物。 Schmid与合作者论述了含饱和脂肪酸键合相的合成与性质,并且与相应的饱和烃固定相的使用作过比较。Pesek和Matyska合成了两种不同的二醇键合相,包括一种是“可靠的”,另一种通过将7-辛烯-1,2-二醇直接连接到氢化硅基质上。Jino和Nakamura [4] 评价了用氟处理的键合硅胶作为一种反相色谱基质与常规反相色谱基质的选择性不同。Buszewki 等比较了烷基胺和烷基键合相基质用于反相分离,发现前者对分离极性化合物效果显著,但其热稳定性不如后者。Wongyai 用苯丙醇胺键合到硅胶上产生离子交换——反相基质的混合模型,并且评价了酸性系列、中性和碱性物质的分离效果。Friebe 将Calixarene键合到硅胶上研究其作为选择相能够形成类似于环糊精的内包合物。Chriswanto 和伙伴们将聚吡咯相涂在硅胶上作为HPLC 的特征化填料。Ge等合成了聚3-辛环吡咯改性硅胶,评价其作为蛋白质的分离及探讨酸、碱介质的稳定性。Skapo和Simpson则在流动床中制得反相基质,他们总结出在有机溶剂中通过这种途径与常规键合相比较可制得重现性更好的基质。Theinpont论述了在HPLC 柱中已经填装好的硅胶的衍生化过程和通过这种途径制得的色谱柱和常规键合相色谱柱进行了比较。1.3 亲和色谱 日本和欧洲较多研究多孔聚合物在液相色谱中的应用,这些聚合物可用于分离各种芳香族化合物和糖类,高分子填料对保留值有较大影响,依据不同溶剂洗脱次序,可估算出聚合物溶解度参数。它们适用于亲水溶质排阻色谱,如有一种吸附剂TSK-SW 硅胶带有亲水键合相,在较大的流速下能经受高压且有非常高的分离度,尤其适用于蛋白质和腐殖酸的分离。曾有人描述过带羟基、氰基、铵离子及芳基金属醚键合到硅胶上,铵离子键合相主要以氢键结合,通过溶剂作用稳定保留值。1.4 离子交换色谱 离子交换固定相的确研究过不少,如八十年代初流行的TSK型SW硅胶及基于二乙胺乙基(DEAE) 制成的阴离子交换材料用于蛋白质及核酸的优化分离,低聚糖的分离采用Micro-Pak AX-5。XAD 树脂是色谱中使用最基本的固定相,弱酸、弱碱及两性电解质的分离均有人研究过,反离子以扩散双电层形式存在。也有人制备多孔甲基丙烯酸盐离子交换剂,但多数人愿意使用商品化的交换剂。Sevec 和Frechet [5] 制得聚甲基丙烯酸缩水甘油酯共聚二甲基乙烯丙烯酸酯色谱柱 (300×8 mm) 用于蛋白质离子交换的制备色谱。他们能做到一次进样到这种色谱柱分离300mg 的蛋白质混合物。Gawdzrk 和 Matynia制备了一种交联的(p,p’—二羟基苯)—丙烷二环氧甘油醚和二乙烯苯的甲基丙烯酸酯的多孔共聚物作为一种最新的HPLC填料并且评价了其作为正相和反相分离的效果。 Danielson 和其合作者[6] 用一种丙胺基硅基质同Kel-F800 反应制得一种弱阴离子交换填料。1.5 空间排阻色谱 Kato 等曾通过丁醚或三乙醇苯醚与TSK G3000 SW 空间排阻色谱法 (SEC) 结合合成 HIC 固定相材料,当使用盐溶液梯度洗脱时,可达到很高的分离度。一些极性键合相通过分配比调节的排阻色谱分离蛋白质和多肽。Miller 等报道过用醚相结构合成大孔硅胶填料:Si-(CH2)3-O-(CH2CH2O)n-R n=1,2或3 R=Me,Et或n-Bu。用这种填料制得的色谱柱可至少连续使用五个月其保留值不变。 烷基链长度和配体密度能直接影响到保留值与分离度,而使用低配位数密度基质更易再生,不易变性,对分离大多数蛋白质来说,基质微球直径在300? 时具有最强的再生力,直径小到1mm的多孔微粒填入1cm 的色谱柱中对于蛋白质的分离十分有用。另一改进色谱有效途径是使用非多孔微球(1-3mm),柱长约30mm,柱效稳定,当小心控制流动相温度和所有仪器组成时,柱外效应很小,曾有人通过洗脱法在2分钟内分离6种蛋白质的混合物。 Smigol[7] 论述了均相聚合甲基丙烯酸缩水甘油酯共聚乙烯二甲基丙烯酸酯微球用特殊化功能的多孔介质制得二醇或二乙胺相有较大孔隙和十八烷基官能团有较少孔隙,这样制得的一种基质当所有分子接近极性相时,较少的分子进入多孔介质,而限制了大分子进入到十八烷基键合相中。 总之,不论是何种类型的固定相对于手性异构体的拆分以及蛋白质和多肽大分子的分离仍在不断的探索完善之中,从发展的趋势看,寻求各种高分子材料作为新型色谱固定相前景光明
高效液相色谱法按组分在固定相和流动相两者间分离机理不同可分为,液固吸附色谱。液液分配色谱,化学键合相色谱法。离子交换色谱法,凝胶色谱法。我疑问的是,我们平时用的ODS-C18柱是固液吸附色谱柱吗???但是好像跟化学键合相色谱也相符合。毕竟色谱柱也是以硅胶为基质键合的C18填料柱。所以我们平时用的这个C18柱是哪一种呢?
请教各位大虾,有谁有固相色谱柱制作流程和技术相关的资料!我想运用这一原理改做其他方面的研究! 或者请发到我的邮箱里也可! wjgsunny@gmail.com QQ:234111734
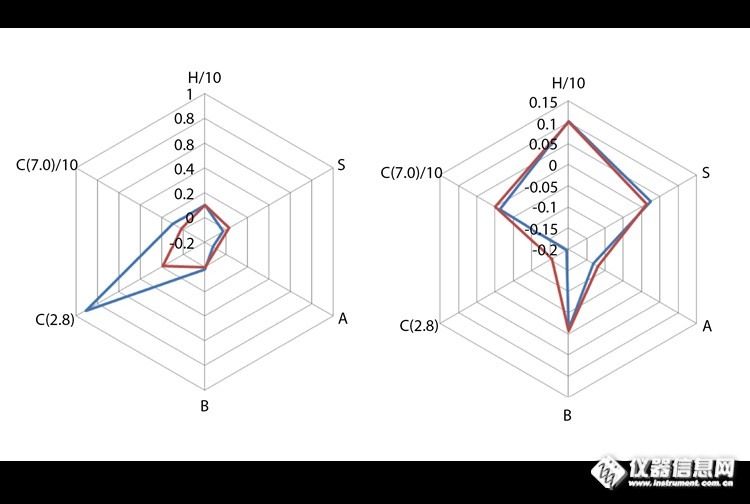
月旭科技与LCGC战略合作,翻译LCGC文章http://ng1.17img.cn/bbsfiles/images/2015/08/201508041725_559105_1863087_3.jpgTony TaylorLCGC North America, Volume 33, Issue 3, page 218 (2015). 反相液相色谱可供选择的固定相种类繁多令人眼花缭乱,即使是某一种固定相(例如C18)的可选择种类也是很多的。 老实说,我们的很多方法开发都是在尝试和错误中进行,这些都是基于我们喜爱的供应商提供的成熟的或者新兴的固定相。即使是先进的含有仔细考量的正交化及电脑优先洗脱设计的“筛选”平台,有时也不得不采取“色谱的本能”。 反相色谱中的保留是基于被分析物、流动相、键合相以及键合了配体的硅胶表面的活性和其可接触性之间的平衡。 想要搞清楚影响分离效果的保留机理,就要考虑并明确化学键合相、活性硅胶表面的处理、硅胶表面的可接触性等因素,这些都将影响色谱柱的原始选择性及方法开发的优化。 在大多数反相分离中色散作用是起主要作用的,尤其是那些使用未改性的烷基配体(C18、C8、C4),其保留能力是与被分析物的疏水性成正比的。含有芳香基团或不饱和基团的固定相或被分析物进行分析时,电荷转移(或π-π)作用是起主要作用的。偶极-氢键相互作用对于极性化合物的保留是很重要的,含有“氰基”的固定相会增强对极性化合物的保留。被分析物的电离部分与硅胶表面之间存在静电作用力,这是由于硅胶表面有残留的可离子化的硅醇基。 当前有许多色谱柱分类系统存在,这些系统都是基于对已知化学探针物质的检测,从而来描述固定相的独特特性。一个非常有用的例子就是美国药典(USP)网站中的产品质量研究数据库(也就是PQRI系统),网址是:http://www.usp.org/app/USPNF/columnsDB.html。该数据库采用保留(1,2)的疏水减法模型来描述固定相的疏水性(H),判断疏水性类似而有不同形状或流体力学体积的被分析物的空间结构选择性参数(S),在pH值为7.0和2.8时的氢键(作为路易斯酸或路易斯碱)和静电作用参数(C)。pH值为7.0时硅醇基活性很强,pH为2.8时具有酸性的硅醇基将会与极性或可离子化的被分析物发生作用产生拖尾。独特的或正交的固定相一般会有较大的S、B和C(7.0)值。这些大型的数据库对于比较固定相的特点是很有用的,“雷达图”也是另一种比较固定相特点的有用方式。表一总结了一些当前常用固定相的分类及其相关应用领域。表一:一些主要固定相分类及其主要应用固定相配体应用C18(烷基)-C18H37大多数烷基固定相可保留药物,类固醇,脂肪酸,邻苯二甲酸酯,环境污染物极性嵌入C18-C16H33NO对氨基甲酸酯及其类似物质等极性强的被分析物保留更强,能够达到更好的分离效果。氰基(氰丙基)-(CH2)3CN对于极性化合物及有多重不同化学成分的溶质有独特的选择性苯基(二苯基)-C6H5(-C12H9)芳香族和中等极性化合物五氟苯基(PFP)-C6F5对于卤化物、极性和异构体物质有较强的选择性氨基(氨丙基)-(CH2)3NH2可用作pH控制的弱阴离子交换剂,用以增强静电保留http://ng1.17img.cn/bbsfiles/images/2015/08/201508050854_559145_1863087_3.png表一:一些常用的反相键合相的保留机理以及键合在硅胶表面的键合相的结构示意图http://ng1.17img.cn/bbsfiles/images/2017/10/2015080417232767_01_1863087_3.png表二:左图是根据PQRI数据库中相似固定相制得的雷达图,右图是根据PQRI数据库中正交固定相制得的雷达图,也就是根据固定相的疏水性来预测其选择性的相似区域。参考文献(1) L.R. Snyder, J.W. Dolan, and P.W. Carr, J. Chromatogr. A 1060, 77–116 (2004).(2) L.R. Snyder, J.W. Dolan, and P.W. Carr, Anal. Chem. 79, 3255–3261 (2007).How to cite this article:Tony Taylor, “The Essentials: Choosing the Right HPLC Stationary Phase,” LCGC North America 33(3), 218 (2015).了解:手性键合相及其应用进展(点击此链接哦!)

手动的固相微萃取装备。用色谱科4毫升样品瓶子装待测样品用完就扔的那种还有更香的推荐吗?原版是这样婶得。。。。。。。。。。。[img=,690,920]https://ng1.17img.cn/bbsfiles/images/2020/05/202005191600194681_1991_2651621_3.jpg!w690x920.jpg[/img]
大家好,刚接触色谱,但想先用固相微萃取发取得样品,然后用GC检测。请问应注意哪方面的知识,特别是在固相微萃取时该注意哪些呢?做完之后怎样清洗,让萃取头可以重复使用?
1.1 正相色谱 八十年代初,人们使用的正相色谱固定相硅胶和吡啶硫氰酸镍盐的络合物晶体能与芳香族化合物形成包合物用于分离芳香族含氮异构体和胆甾醇晶体。已有人[1]将焦炭吸附剂作为填料和键合硅胶作过比较并研究其热力学机理。具有离子化或非离子化功能团的大孔聚合物也开始应用于液相色谱,这些聚合物在整个酸碱范围内稳定,其中一个缺点是当溶剂改变时,它们会热胀冷缩,但是其主要的分析方面用途,即从水中收集痕量研究化合物是没有问题的,通过适当的处理装入色谱柱,一些物质出现过度的峰带变宽 ,而另一些则出现尖、窄的峰带和高分离度,在强碱性大孔树脂中120分钟内可分离100 种尿液中紫外吸收物质[2]。有人将各种链长度的碳氢配位体键合到硅胶上,根据链长短效应研究保留值的影响,也有过类似的报道,将C22 键合相以及制备键合相担体各种条件作过比较,其键长度和吸附自由能成线性关系。一般地,碱溶液会破坏硅胶,烷基胺比季铵盐更会腐蚀硅胶填料,故通过加填有5m m 硅胶的短预柱来洗脱碱性溶液。Kataev等[3]用聚三氟苯乙烯涂成的硅胶微粒基质并应用到卤代芳烃、多肽和蛋白质的分离。1993年11月,Majors 讨论了在日本发展迅速的HPLC 聚合物填料。Hosoya 制备了大孔聚合乙烯—对—叔丁基苯甲酸丁酯微球和两种别的聚合物微球的性能以及更多的C18HPLC固定相。 1.2 反相色谱 固定相的研究进入九十年代更是热火朝天。这方面的研究报道远远超过流动相的研究,人们不断探索保留过程及相关烷基键结构的复杂性和可能制得的各种新型固定相。Bolok 和其合作者从统计技术研究反相色谱柱的老化过程。Montes 等评价了一种硅氢化物介质的硅氢烯制备烷基键合相,更早时候基质制备可供参考。Moriyama等评价了用2mm硅胶制得的TSK胶Super ODS新型反相色谱柱的特性,他们论述了如何分离手性化合物。 Schmid与合作者论述了含饱和脂肪酸键合相的合成与性质,并且与相应的饱和烃固定相的使用作过比较。 Pesek和Matyska合成了两种不同的二醇键合相,包括一种是“可靠的”,另一种通过将7-辛烯-1,2-二醇直接连接到氢化硅基质上。Jino和Nakamura [4] 评价了用氟处理的键合硅胶作为一种反相色谱基质与常规反相色谱基质的选择性不同。Buszewki 等比较了烷基胺和烷基键合相基质用于反相分离,发现前者对分离极性化合物效果显著,但其热稳定性不如后者。Wongyai 用苯丙醇胺键合到硅胶上产生离子交换——反相基质的混合模型,并且评价了酸性系列、中性和碱性物质的分离效果。Friebe 将Calixarene键合到硅胶上研究其作为选择相能够形成类似于环糊精的内包合物。Chriswanto 和伙伴们将聚吡咯相涂在硅胶上作为HPLC 的特征化填料。Ge等合成了聚3-辛环吡咯改性硅胶,评价其作为蛋白质的分离及探讨酸、碱介质的稳定性。Skapo和Simpson则在流动床中制得反相基质,他们总结出在有机溶剂中通过这种途径与常规键合相比较可制得重现性更好的基质。Theinpont 论述了在HPLC 柱中已经填装好的硅胶的衍生化过程和通过这种途径制得的色谱柱和常规键合相色谱柱进行了比较。 1.3 亲和色谱 日本和欧洲较多研究多孔聚合物在液相色谱中的应用,这些聚合物可用于分离各种芳香族化合物和糖类,高分子填料对保留值有较大影响,依据不同溶剂洗脱次序,可估算出聚合物溶解度参数。它们适用于亲水溶质排阻色谱,如有一种吸附剂TSK-SW 硅胶带有亲水键合相,在较大的流速下能经受高压且有非常高的分离度,尤其适用于蛋白质和腐殖酸的分离。曾有人描述过带羟基、氰基、铵离子及芳基金属醚键合到硅胶上,铵离子键合相主要以氢键结合,通过溶剂作用稳定保留值。 1.4 离子交换色谱 离子交换固定相的确研究过不少,如八十年代初流行的TSK型SW硅胶及基于二乙胺乙基(DEAE) 制成的阴离子交换材料用于蛋白质及核酸的优化分离,低聚糖的分离采用Micro-Pak AX-5。XAD 树脂是色谱中使用最基本的固定相,弱酸、弱碱及两性电解质的分离均有人研究过,反离子以扩散双电层形式存在。也有人制备多孔甲基丙烯酸盐离子交换剂,但多数人愿意使用商品化的交换剂。Sevec 和Frechet [5] 制得聚甲基丙烯酸缩水甘油酯共聚二甲基乙烯丙烯酸酯色谱柱 (300×8 mm) 用于蛋白质离子交换的制备色谱。他们能做到一次进样到这种色谱柱分离300mg 的蛋白质混合物。Gawdzrk 和 Matynia制备了一种交联的(p,p’—二羟基苯)—丙烷二环氧甘油醚和二乙烯苯的甲基丙烯酸酯的多孔共聚物作为一种最新的HPLC填料并且评价了其作为正相和反相分离的效果。 Danielson 和其合作者[6] 用一种丙胺基硅基质同Kel-F800 反应制得一种弱阴离子交换填料。 1.5 空间排阻色谱 Kato 等曾通过丁醚或三乙醇苯醚与TSK G3000 SW 空间排阻色谱法 (SEC) 结合合成 HIC 固定相材料,当使用盐溶液梯度洗脱时,可达到很高的分离度。一些极性键合相通过分配比调节的排阻色谱分离蛋白质和多肽。Miller 等报道过用醚相结构合成大孔硅胶填料:Si-(CH2)3-O-(CH2CH2O)n-R n=1,2或3 R=Me,Et或n-Bu。用这种填料制得的色谱柱可至少连续使用五个月其保留值不变。 烷基链长度和配体密度能直接影响到保留值与分离度,而使用低配位数密度基质更易再生,不易变性,对分离大多数蛋白质来说,基质微球直径在300? 时具有最强的再生力,直径小到1mm的多孔微粒填入1cm 的色谱柱中对于蛋白质的分离十分有用。另一改进色谱有效途径是使用非多孔微球(1-3mm), 柱长约30mm,柱效稳定,当小心控制流动相温度和所有仪器组成时,柱外效应很小,曾有人通过洗脱法在2分钟内分离6种蛋白质的混合物。 Smigol[7] 论述了均相聚合甲基丙烯酸缩水甘油酯共聚乙烯二甲基丙烯酸酯微球用特殊化功能的多孔介质制得二醇或二乙胺相有较大孔隙和十八烷基官能团有较少孔隙,这样制得的一种基质当所有分子接近极性相时,较少的分子进入多孔介质,而限制了大分子进入到十八烷基键合相中。 总之,不论是何种类型的固定相对于手性异构体的拆分以及蛋白质和多肽大分子的分离仍在不断的探索完善之中,从发展的趋势看,寻求各种高分子材料作为新型色谱固定相前景光明。
[b]有奖问答’选择题: 当色谱分析的流动相是液体, 固定相是固体时. 该色谱称为( ) (A) 气-固色譜 (B) 气-液色譜 (C) 液-固色譜 (D) 液-液色譜[/b]
细管柱是气固色谱柱还是气液色谱柱,如何从固相判断其极性?[em01]

C1。 反相液相色谱的固定相选择有很多种,即使特定的固定相(如C18)其选择的种类也非常多。在反相液相色谱的固定相选择时,要考虑化学键合相、活性硅胶表面的处理、硅胶表面的可接触性等,这些因素都会影响到液相色谱最后的分离效果以及方法开发的优化。 分离中等极性和极性较强的化合物可选择极性键合相。氨基键合相具有较强的氢键结合能力,对某些多官能团化合物如甾体、强心甙等有较好的分离能力;氨基键合相上的氨基能与糖类分子中的羟基产生选择性相互作用,故被广泛用于糖类的分析,但它不能用于分离羰基化合物,如甾酮、还原糖等,因为它们之间会发生反应生成Schiff 碱。分离极性较弱和非极性的化合物可选择非极性键合相。非极性键合相也可用于分离离子型或可离子化的化合物。ODS(octadecyl silane)是应用最为广泛的非极性键合相,它对各种类型的化合物都有很强的适应能力。短链烷基键合相能用于极性化合物的分离,而苯基键合相适用于分离芳香化合物。 色散作用在许多反相分离中都发挥着重要的作用,特别是在未改性的烷基配体(C18、C8、C4)中,其保留能力与被分析物的疏水性成正比。固定相或被分析物在含有芳香基团或不饱和基团时,电荷转移(或π-π)起重要的作用。当被分析物为极性化合物时,含有“氰基”的固定相会增强对极性化合物的保留,这是因为固定相硅胶表面有离子化的硅醇基与被分析物之间存在静电作用力。 表一总结了一些当前常用固定相的分类及其相关应用领域。表一:一些主要固定相分类及其主要应用 固定相 配体 应用 C18(烷基) -C18H37 大多数烷基固定相可保留药物,类固醇,脂肪酸,邻苯二甲酸酯,环境污染物 极性嵌入C18 -C16H33NO 对氨基甲酸酯及其类似物质等极性强的被分析物保留更强,能够达到更好的分离效果。 氰基(氰丙基) -(CH2)3CN 对于极性化合物及有多重不同化学成分的溶质有独特的选择性 苯基(二苯基) -C6H5(-C12H9) 芳香族和中等极性化合物 五氟苯基(PFP) -C6F5 对于卤化物、极性和异构体物质有较强的选择性 氨基(氨丙基) -(CH2)3NH2 可用作pH控制的弱阴离子交换剂,用以增强静电保留http://ng1.17img.cn/bbsfiles/images/2015/08/201508281618_563401_3005330_3.jpg图一、一些常用的反相键合相的保留机理以及键合在硅胶表面的键合相的结构示意图
最近想要用高效液相色谱-固相微萃取做点东西,可是没有什么进展,麻烦各位老师指点迷津!谢谢! 我尝试了做不同的样品,可基本上是没检出什么东西来。 1.黄胺类药物残留,用的是PA和PDMS纤维头,萃取30分钟,静态解吸3min,基本只有比燥声大一点的峰。 2.三聚氰胺 3.黄曲霉毒素 都以失败告终。 最后我就直接按人家发表的文献重做了一下氨基甲酸酯农药,结果还是很不如人意啊! 请问用高效液相色谱-固相微萃取是不是所有用液相能检测的物质都能用固相微萃取来检测吗?特别是残留,还有做的时候在最要注意的是什么?
第七课 液相色谱仪 [url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]法是一种很好的分离、分析方法,它具有分析 速度快、分离效能好和灵 敏度高等优点。但是[url=https://insevent.instrument.com.cn/t/Mp]气相[/url]色 谱仅能分析在操作温度下能汽化而不分解的物质。据估 计,在已知化合物中能直接进行[url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]分析的化合物 约占15%,加上制成衍生物的化合物,也不过20%左右。 对于高沸点化合物;难挥发及热不稳定的化合物、离子 型化合物及高聚物等,很难用[url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]法分析。为解决 这个问题,70年代初发展了高效液相色谱。高效液相色 谱的原理与经典液相色谱相同,但是它采用了高效色谱 拄、高压泵和高灵敏度检测器。因此,高效液相色谱的 分离效率、分析速度和灵敏度大大提高。 就其分离机 理的不同,高效液相色谱可以分为液-固吸附色谱、 液-液分配色谱、 离子交换色谱和凝胶渗透色谱四类。 液—固色谱的色谱柱内填充固体吸附剂,由于不同组分 具有不同的吸附能力,因此, 流动相带着被测组分经 过色谱柱时,各组分被分开。 液—液色谱的流动相和 固定相都是液体。作为固定相的液体涂在惰性担体上, 流动 相与固定液不互溶。当带有被测组分的流动相进 入色谱柱时,组分在两相间很快达分配平衡,由于各 组分在两相间分配系数不同而彼此分离。以非极性溶 液作流动相,极性物质作固定相的液—液色谱叫正相 色谱;极性溶液作流动相,非极性物质作固定相的液 —液色谱叫反相色谱。 离子交换色谱的色谱柱内填充 离子交换树脂,依靠样品离子交换能力的差别实现分 离。而凝胶色谱是按试样中分子大小的不同来进行分 离的。 在上述四类色谱中,应用最广泛的是液—液色 谱,因此,在本节的讨论中以液—液 色谱为主。 高 效液相色谱的基本理论和定性定量分析方法与[url=https://insevent.instrument.com.cn/t/Mp]气相[/url]色 谱基本相同。高效液相色谱仪由输液系统、进样系统、 分离系统、检测系统和数据处理系统组成。
第九课 液相色谱仪-进样系统,分离系统 进样系统一般高效液相色谱多采用六通阀进样。先由注射器将样 品常压下注入样品环。然后 切换阀门到进样位置,由 高压泵输送的流动相将样品送人色谱柱。样品环的容积 是固定的,因此进样重复性好。 分离系统 分离系统包括色谱柱、连接管、恒温器等。色谱柱是高 效液相色谱仪的心脏。它是 由内部抛光的不锈钢管制成 ,一般长10—50cm,内径2—5mm,柱内装有固定相。液 相色谱的固定相是将固定该涂在担体上而成。担体有两 类:一类是表面多孔型担体;另一类是全多孔型担体。 近年来又出现了全多孔型微粒担体。这种担体检度为5 —10 um,是由nm级的硅胶微粒堆积而成,又叫堆积硅 珠。由于颗粒小,所以柱效高,是目前最广泛使用的一 种担体。 在高效液相色谱分析中,适当提高柱温可改善 传质,提高桂效,缩短分析时间。因 此,在分析时可以 采用带有恒温加热系统的金属夹套来保持色谱拄的温度。 温度可以在室温到60℃间调节。
目标物分子从[url=https://insevent.instrument.com.cn/t/5p][color=#3333ff]液相色谱[/color][/url]流动相转移进入固定相或从固定相移出重新进入流动相过程,会引起色谱峰形的明显扩散。流动相中目标物分子的迁移速度依赖于它在液固色谱的固相上的吸附和解吸,当分子被吸附在吸附剂活性作用点上时,它再从表面解吸会有较大阻力,当它最后解吸时必然会落在已随载液前行的大部分分子之后。
[b]液相色谱固定相探索完善与发展[/b]1.1 正相色谱 八十年代初,人们使用的正相色谱固定相硅胶和吡啶硫氰酸镍盐的络合物晶体能与芳香族化合物形成包合物用于分离芳香族含氮异构体和胆甾醇晶体。已有人[1]将焦炭吸附剂作为填料和键合硅胶作过比较并研究其热力学机理。具有离子化或非离子化功能团的大孔聚合物也开始应用于液相色谱,这些聚合物在整个酸碱范围内稳定,其中一个缺点是当溶剂改变时,它们会热胀冷缩,但是其主要的分析方面用途,即从水中收集痕量研究化合物是没有问题的,通过适当的处理装入色谱柱,一些物质出现过度的峰带变宽 ,而另一些则出现尖、窄的峰带和高分离度,在强碱性大孔树脂中120分钟内可分离100 种尿液中紫外吸收物质[2]。有人将各种链长度的碳氢配位体键合到硅胶上,根据链长短效应研究保留值的影响,也有过类似的报道,将C22 键合相以及制备键合相担体各种条件作过比较,其键长度和吸附自由能成线性关系。一般地,碱溶液会破坏硅胶,烷基胺比季铵盐更会腐蚀硅胶填料,故通过加填有5m m 硅胶的短预柱来洗脱碱性溶液。Kataev等[3]用聚三氟苯乙烯涂成的硅胶微粒基质并应用到卤代芳烃、多肽和蛋白质的分离。1993年11月,Majors 讨论了在日本发展迅速的HPLC 聚合物填料。Hosoya 制备了大孔聚合乙烯—对—叔丁基苯甲酸丁酯微球和两种别的聚合物微球的性能以及更多的C18HPLC固定相。 1.2 反相色谱 固定相的研究进入九十年代更是热火朝天。这方面的研究报道远远超过流动相的研究,人们不断探索保留过程及相关烷基键结构的复杂性和可能制得的各种新型固定相。Bolok 和其合作者从统计技术研究反相色谱柱的老化过程。Montes 等评价了一种硅氢化物介质的硅氢烯制备烷基键合相,更早时候基质制备可供参考。Moriyama等评价了用2mm硅胶制得的TSK胶Super ODS新型反相色谱柱的特性,他们论述了如何分离手性化合物。 Schmid与合作者论述了含饱和脂肪酸键合相的合成与性质,并且与相应的饱和烃固定相的使用作过比较。 Pesek和Matyska合成了两种不同的二醇键合相,包括一种是“可靠的”,另一种通过将7-辛烯-1,2-二醇直接连接到氢化硅基质上。Jino和Nakamura [4] 评价了用氟处理的键合硅胶作为一种反相色谱基质与常规反相色谱基质的选择性不同。Buszewki 等比较了烷基胺和烷基键合相基质用于反相分离,发现前者对分离极性化合物效果显著,但其热稳定性不如后者。Wongyai 用苯丙醇胺键合到硅胶上产生离子交换——反相基质的混合模型,并且评价了酸性系列、中性和碱性物质的分离效果。Friebe 将Calixarene键合到硅胶上研究其作为选择相能够形成类似于环糊精的内包合物。Chriswanto 和伙伴们将聚吡咯相涂在硅胶上作为HPLC 的特征化填料。Ge等合成了聚3-辛环吡咯改性硅胶,评价其作为蛋白质的分离及探讨酸、碱介质的稳定性。Skapo和Simpson则在流动床中制得反相基质,他们总结出在有机溶剂中通过这种途径与常规键合相比较可制得重现性更好的基质。Theinpont 论述了在HPLC 柱中已经填装好的硅胶的衍生化过程和通过这种途径制得的色谱柱和常规键合相色谱柱进行了比较。来源:中国色谱网。
请问有人用过在线固相萃取液相色谱仪吗?上面用的固相萃取柱是不是就是截短的普通色谱柱?能不能用保护柱来代替?还是说对填料粒径有要求,太小容易堵,一般在10-20um?一般用什么品牌的SPE柱比较多?Waters的一根HLB材质的Online-SPE柱好贵,要五六千,用不起,有便宜点的SPE柱品牌推荐吗?
高效液相色谱手性固定相研究进展 作者:寿崇琦,张志良,赵春宾,邢希学,李关宾,陈立仁摘要:对近年来高效液相色谱手性固定相的研究进行了综述。重点介绍了手性固定相的分类、拆分机理和应用的新进展。讨论了各类手性固定相优缺点,提出了目前存在的问题、今后的研究方向和重点。 关键词:高效液相色谱;手性固定相;拆分机理 随着生物工程和生物科学的发展,手性拆分和测定引起了人们的普遍关注。尽管对映体间物理化学性质几乎完全相同,但它们的生化和药理作用却往往不同。这是因为生物本身内部的核酸、蛋白质及多糖都具有与其功能相适应的结构,它们常常对扬长避短一化合物的两种对映体表现出不同的响应。例如具有镇静作用的反应停(thalidomide,酞胺哌啶酮),其有效成分是R构型,而S构型则具有致畸作用⋯ 。据统计,常用的2OO种药物中,大约有120种至少含有一个手性中心。而这些手性药物中有80%~90%以外消旋体形式在市场销售,存在巨大的潜在危险性。因此,对映体的拆分与识别对于生命科学和药物化学研究以及人类的健康具有十分重要的意义。 目前用于手性分离的方法主要有毛细管电泳法、薄层色谱法、亚临界及超临界流体色谱法、[url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]法和液相色谱法。近年来,高效液相色谱法取得了令人瞩目的进展,已成为对映体拆分强有力的手段之一。而其中所用的手性固定相的是能否进行手性分离的关键。 l 手性固定相的分类 虽然液相色谱常被分为不同的分离模式,但实质上所有的分离模式都基于两个最基本的因素:即固定相的结构和组成,以及决定分离机理的固定相与流动相相互作用的性质。因而手性固定相(CSP)的制备则是手性分离的关键。目前所研究的HPLC-CSP主要可分为下列几类: 1.1 蛋白质手性亲和固定相 多数蛋白质CSP的分离机理目前尚不十分清楚,但是蛋白质CSP的手性识别能力可以归结为它们独特的空间立体结构特征。尤其是在对映体的手性识别过程中,三级结构所造成的疏水性口袋、沟槽或通道对手性拆分具有十分重要的意义。 七十年代初已有人将牛血清白蛋白键合在琼脂糖上,制成液相色谱用手性填料,拆分了DL-色胺酸。八十年代初,Alhnmark和Hellnanssor分别将BSA和a-酸性糖蛋(a-AGP)键合到微粒硅胶上制成HPLC用的CSP,商品名为Resolvosil和Enantio Pac。Hermanssor和Schill等以Enantio Pac柱拆分了酸、胺和β-氨基醇类药物如萘普生、双异丙吡胺、麻黄素、可卡因、阿托平等几十种药物,拆分效果良好,分离系数(a)可达1.1~4.0,柱的稳定性好,对温度和有机溶剂有较好的耐受性,长期保存在异丙醇中(12个月),对拆分效果无明显影响。Resolvosil柱可用于拆分氨基及其衍生物、芳香亚砜、二苯乙醇及多种药如安定、华法林等。在国内,张鹏等用万古霉素制备的手性固定相用于对布洛芬手性对映体的拆分,也取得了不错的效果。周华等用L-脯氨酸衍生的衍生物作为高效液相色谱手性固定相,用于对4种N-3,5-二硝基苯甲酰-DL-氨基酸甲酯对映体色谱拆分,结果显示对映体选择性在1.04一1.18之间。兰州化学物理研究所的候经国在这一方面也做了深入研究。 1.2 环糊精型手性固定相 手性固定相拆分对映体的另一途径是将手性空穴键合到固定相表面,由此产生的客体-受体间的相互作用决定对映体的拆分效果。最常用的是环糊精手性固定相,环糊精手性固定相的分离机理主要基于包含型络合过程,这一机理表示分子的非极性部分被吸引到非极性空穴中,当被分离的分子存在芳香基团时,由于芳香基团与糖苷键上氧原子的作用产生立体选择的趋向,而线性或无环的烷烃以随机方式占据环糊精的空穴。 Ward将β-CD通过6~10碳的间隔基键合到微粒硅胶上,商品名Cyclobond I。这种CSP象切去顶端的锥形圆筒,边缘有羟基,内部为疏水性的内腔,只有当分子的大小正好使非极性部分进入腔内,极性基团与边缘的羟基产生较强的作用才能拆分。某些具有萘环和双环化合物可得到良好的拆分,如丹酰氨基酸、巴比妥类、乙内酰脲、金属茂等。单环化合物如扁桃酸、酪氨酸、麻黄素等也可拆分,使用范围广,仅次于Pirkle型GSP。流动相为甲醇-水或乙睛-水,γ-CD无拆分作用,a-CD仅对小分子有拆分作用。唐课文等合成了一种烷基化和硅烷化β-环糊糖手性固定相,用其分离了醇、酮、烯烃等一些对映体,取得了很好的效果。辛梅华等人将β-环糊精进行了乙酰化用于对肾上腺素类对映体的测定,也得到了不错的结果。 1.3 配体交换型固定相 这类手性固定相拆分对映体与手性配体流动相的拆分机理相同,只是手性配体键合到固定相基质成为手性固定相而已。其配体分子亦为具有手性活性的氨基酸如脯氨酸,金属离子多用Cu2+。Davank0v将L-脯氨酸键合到树脂上,经吸附CU(Ⅱ)、Ni(Ⅱ)或Zn(Ⅱ)后,可拆分氨基酸对映体。Gubity将L-脯氨酸和L-缬氨酸通过间隔基键合到硅胶上,制成HPLC用CSP,以CuSO4水溶液处理后即可用于氨基酸和二肽类的拆分。此类CSP已有四种商品:Chiracel WH,WM和Chiral hypro-Cu,Val-Cu。除上述氨基酸外,用为CSP键合基的尚有:L-l,2-二氨丙烷、β-羟基氨基酸、l-麻黄素、L-组氨酸、L-苯丙氨酸、L-氮杂-2-环丁烷羧酸、L-酒石酸、L-2-哌啶酸、及(一)-反-1,2-环已二胺。拆分化合物包括氨基酸、二肽、a-羟基酸及有关药物如a-甲基多巴、甲状腺激素等,a值1.10—8.0,配位基交换色谱的流动相多为水或磷酸、乙酸氨缓冲液,有时加入一定量的有机溶剂(乙腈)。为防止铜离子的流失,在流动相中加入适当的CuSO4,此类CSP亦可用于制备性分离。
高效液相色谱手性固定相研究进展摘要:对近年来高效液相色谱手性固定相的研究进行了综述。重点介绍了手性固定相的分类、拆分机理和应用的新进展。讨论了各类手性固定相优缺点,提出了目前存在的问题、今后的研究方向和重点。 关键词:高效液相色谱;手性固定相;拆分机理
在色谱分析中,如何选择最佳的色谱条件以实现最理想分离,是色谱工作者的重要工作,也是用计算机实现HPLC分析方法建立和优化的任务之一。本章着重讨论填料基质、化学键合固定相和流动相的性质及其选择。一、基质(担体) HPLC填料可以是陶瓷性质的无机物基质,也可以是有机聚合物基质。无机物基质主要是硅胶和氧化铝。无机物基质刚性大,在溶剂中不容易膨胀。有机聚合物基质主要有交联苯乙烯-二乙烯苯、聚甲基丙烯酸酯。有机聚合物基质刚性小、易压缩,溶剂或溶质容易渗入有机基质中,导致填料颗粒膨胀,结果减少传质,最终使柱效降低。1.基质的种类1)硅胶 硅胶是HPLC填料中最普遍的基质。除具有高强度外,还提供一个表面,可以通过成熟的硅烷化技术键合上各种配基,制成反相、离子交换、疏水作用、亲水作用或分子排阻色谱用填料。硅胶基质填料适用于广泛的极性和非极性溶剂。缺点是在碱性水溶性流动相中不稳定。通常,硅胶基质的填料推荐的常规分析pH范围为2~8。硅胶的主要性能参数有:①平均粒度及其分布。②平均孔径及其分布。与比表面积成反比。③比表面积。在液固吸附色谱法中,硅胶的比表面积越大,溶质的k值越大。④含碳量及表面覆盖度(率)。在反相色谱法中,含碳量越大,溶质的k值越大。⑤含水量及表面活性。在液固吸附色谱法中,硅胶的含水量越小,其表面硅醇基的活性越强,对溶质的吸附作用越大。⑥端基封尾。在反相色谱法中,主要影响碱性化合物的峰形。⑦几何形状。硅胶可分为无定形全多孔硅胶和球形全多孔硅胶,前者价格较便宜,缺点是涡流扩散项及柱渗透性差;后者无此缺点。⑧硅胶纯度。对称柱填料使用高纯度硅胶,柱效高,寿命长,碱性成份不拖尾。2)氧化铝 具有与硅胶相同的良好物理性质,也能耐较大的pH范围。它也是刚性的,不会在溶剂中收缩或膨胀。但与硅胶不同的是,氧化铝键合相在水性流动相中不稳定。不过现在已经出现了在水相中稳定的氧化铝键合相,并显示出优秀的pH稳定性。3)聚合物 以高交联度的苯乙烯-二乙烯苯或聚甲基丙烯酸酯为基质的填料是用于普通压力下的HPLC,它们的压力限度比无机填料低。苯乙烯-二乙烯苯基质疏水性强。使用任何流动相,在整个pH范围内稳定,可以用NaOH或强碱来清洗色谱柱。甲基丙烯酸酯基质本质上比苯乙烯-二乙烯苯疏水性更强,但它可以通过适当的功能基修饰变成亲水性的。这种基质不如苯乙烯-二乙烯苯那样耐酸碱,但也可以承受在pH13下反复冲洗。 所有聚合物基质在流动相发生变化时都会出现膨胀或收缩。用于HPLC的高交联度聚合物填料,其膨胀和收缩要有限制。溶剂或小分子容易渗入聚合物基质中,因为小分子在聚合物基质中的传质比在陶瓷性基质中慢,所以造成小分子在这种基质中柱效低。对于大分子像蛋白质或合成的高聚物,聚合物基质的效能比得上陶瓷性基质。因此,聚合物基质广泛用于分离大分子物质。2.基质的选择 硅胶基质的填料被用于大部分的HPLC分析,尤其是小分子量的被分析物,聚合物填料用于大分子量的被分析物质,主要用来制成分子排阻和离子交换柱。二、化学键合固定相 将有机官能团通过化学反应共价键合到硅胶表面的游离羟基上而形成的固定相称为化学键合相。这类固定相的突出特点是耐溶剂冲洗,并且可以通过改变键合相有机官能团的类型来改变分离的选择性。1.键合相的性质 目前,化学键合相广泛采用微粒多孔硅胶为基体,用烷烃二甲基氯硅烷或烷氧基硅烷与硅胶表面的游离硅醇基反应,形成Si-O-Si-C键形的单分子膜而制得。硅胶表面的硅醇基密度约为5个/nm2,由于空间位阻效应(不可能将较大的有机官能团键合到全部硅醇基上)和其它因素的影响,使得大约有40~50%的硅醇基未反应。 残余的硅醇基对键合相的性能有很大影响,特别是对非极性键合相,它可以减小键合相表面的疏水性,对极性溶质(特别是碱性化合物)产生次级化学吸附,从而使保留机制复杂化(使溶质在两相间的平衡速度减慢,降低了键合相填料的稳定性。结果使碱性组分的峰形拖尾)。为尽量减少残余硅醇基,一般在键合反应后,要用三甲基氯硅烷(TMCS)等进行钝化处理,称封端(或称封尾、封顶,end-capping),以提高键合相的稳定性。另一方面,也有些ODS填料是不封尾的,以使其与水系流动相有更好的"湿润"性能。 由于不同生产厂家所用的硅胶、硅烷化试剂和反应条件不同,因此具有相同键合基团的键合相,其表面有机官能团的键合量往往差别很大,使其产品性能有很大的不同。键合相的键合量常用含碳量(C%)来表示,也可以用覆盖度来表示。所谓覆盖度是指参与反应的硅醇基数目占硅胶表面硅醇基总数的比例。 pH值对以硅胶为基质的键合相的稳定性有很大的影响,一般来说,硅胶键合相应在pH=2~8的介质中使用。
高效液相色谱常见问题与解答1 何谓反相柱、正相柱?答:“反相”和“正相”的概念是液相色谱法早期提出的概念,当时键合相色谱柱尚未出现,固定相被涂覆在载体表面,极易流失,为此科学家对流动相使用给出了合理的建议:流动相极性与固定液极性应具有较大差别,以减少固定液流失。固定相极性弱于流动相时的液相色谱法被称为反相色谱法,固定相极性强于流动相时的液相色谱法被称为正相色谱法。尽管目前键合相色谱柱已成为主流,但这一概念在色谱方法开发、预测出峰顺序等方面具有重要意义。由上面的介绍可知具体的色谱方法、色谱柱属于正相还是反相不仅取决于固定相极性,同时还取决于流动相极性。C18(硅胶键合十八烷基硅烷)、C8(硅胶键合辛基硅烷)、PH(硅胶键合苯基硅烷)等色谱柱,由于固定相极性极低,比目前已知的任何流动相的极性都要低,因而是标准的反相柱;Silica(硅胶)、NH2(硅胶键合氨丙基硅烷)具有较高的极性,主要用于分离带有极性基团的化合物,所用流动相的极性通常低于这些固定相,因而是标准的正相柱。CN(硅胶键合腈丙基)的极性适中,当流动相极性超过CN时,它属于反相柱,反之则是正相柱。2 色谱柱规格对分析结果会产生何种影响?答:色谱柱内径决定载样量,载样量与内径的平方成正比;色谱柱长度与塔板数成正比,与柱压成正比;粒径影响涡流扩散相,粒径越小涡流扩散相越小,柱效越高,粒径与柱效近似成反比;粒径越小,压力也越大,压力与粒径的平方成反比。填料孔径对分析对象的分子量有限制,当孔径为分析物尺寸的5倍以上时,分析物才能顺利通过孔隙,孔径处于60~120 Å的色谱柱适用于相对分子量小于10000的分析物,孔径为300 Å的色谱柱可以满足分子量处于10000以上的大分子化合物分析。3 液相色谱分析中如何才能提高分离度?答:下式为分离度计算公式http://www.dikma.com.cn/Public/Uploads/images/R.JPGN:柱效(Efficiency)反映色谱柱性能,柱效越高,分离度越好。在其他条件恒定的情况下,塔板数增加一倍,分离度仅提高40%。操作中,可通过下面两种方式增加塔板数进而提高分离度:其一,使用长柱或双柱串联,但也会使分离时间大大延长;其二,使用细粒径填料的色谱柱,但这需要耐更高压力的液相色谱系统。相比之下后者更为可取。α:选择性(selectivity)是指色谱柱-流动相体系分离两个化合物的能力。选择性主要与固定相、流动相组成以及柱温等因素有关,与保留值也密切相关,其中固定相和流动相组成影响较大。以最常见的反相模式为例,反相柱(包括C18、C8、PH等)是以分配作用对化合物进行保留的,不同化合物的分离是基于它们在键合相与流动相中分配系数的差异,如果两种化合物的水溶性、在烷烃-水体系的分配系数等方面存在明显差异,那么这些化合物通常是能够利用反相柱达到分离;PH柱对具有苯环的化合物具有特殊保留。正相模式下,硅胶柱、胺基柱、氰基柱与带有极性基团的化合物之间存在极性相互作用,对化合物的基团具有选择性,常常用于结构类似物、异构体化合物的分离。流动相方面,降低流动相的洗脱强度通常可以增大分离度;而有机溶剂类型也会影响分离,比如反相条件下,乙腈和甲醇的选择性就存在很大差异,这种差异需要在实践中摸索,但无论如何,多种溶剂类型带给我们更多的实现分离的可能。k:随着容量因子k的增大,分离度也随之增加,这种影响在k值较低时非常明显,当k值大于10时,k值增加对分离度的影响就不再显著,这就告诫无原则地提高k值以增大分离度是没有意义的。增加键合相密度能够提高k值;另外改变键合基团类型也能改变k值,比如在反相色谱中,随着键合相碳链长度的增加,k值逐渐增大。4 色谱峰的峰形是怎样衡量的?有何规定?理论上讲,色谱峰应符合高斯曲线分布,然而实际上任何色谱峰都对高斯曲线分布存在一定的偏离,亦即不对称。峰拖尾可以用不对称因子(As)或USP拖尾因子(Tf)来衡量,显然不对称因子的说法更准确,因为色谱峰存在前延、完美对称、拖尾三种形态。一般来说,制药行业以USP拖尾因子作为评测标准,而其他行业则多采用As来测定峰形。http://www.dikma.com.cn/Public/Uploads/images/tuoweiyinzi.JPG对于药物分析,通常有明确的规定,Tf应处于某一范围之间,比如我国药典规定某些药物的拖尾因子应处于0.95~1.05之间。其他行业尚无较为明确的规定。5 什么是梯度洗脱?什么情况下使用梯度洗脱?答:为了改善分析结果,某些操作需要连续改变流动相中各溶剂组分的比例以连续改变流动相的极性,使每个分析组分都有合适的容量因子k,并使样品种的所有组分可在最短时间内实现最佳分离,这种洗脱方式称为梯度洗脱。梯度洗脱可在下列情形中发挥重要作用:A 在等度下具有较宽k值的多种样品分析。B 大分子样品分析。C 样品含有强保留的干扰物,在目标化合物出峰后设置梯度洗脱,将干扰物洗脱出来,以免其影响下一次数据采集。D 分析方法建立时,不知道其洗脱情况,使用梯度洗脱,找出其较优的洗脱条件。6 何谓封端?封端的意义是什么?硅胶表面的硅羟基(Si-OH)密度为8 μmol/m2,由于空间位阻的存在,硅烷化键合反应最多只能覆盖50%的硅羟基,超过一半的硅羟基是活性硅羟基。这些硅羟基与化合物的极性基团存在极性相互作用和离子交换作用,为化合物的保留增加了不被期望的作用力,往往会影响峰形。用短链氯硅烷(如三甲基氯硅烷)键合活性的硅羟基,可以减小甚至消除这种影响,这种操作被称为封端(Endcap)。封端处理的意义:抑制了特异性吸附,提高了色谱峰的对称性,并改善了分离效果;在一定程度上掩蔽了硅胶表面,使其对碱性环境的耐受性增强;通过空间位阻掩盖了键合反应形成的Si-O-Si键,使其对酸性环境的耐受性增强;可能会影响对极性样品的选择性。7 柱床塌陷和键合相塌陷柱床塌陷是指色谱柱使用一段时间后色谱柱入口处的柱床产生可见的空隙。该空隙的存在增大了死体积,会导致色谱柱柱效下降。造成柱床塌陷的原因如下:其一,色谱柱填装时的压力过低,填装不紧密,在高压下使用一段时间,开始出现空隙;其二,操作压力超出色谱柱填料的耐压值,导致填料颗粒破碎产生空隙;其三,流动相溶解填料导致空隙出现。键合相塌陷是指由于流动相极性与键合相极性相差太大,键合相无法在流动相中充分伸展而倒伏、缠结在一起,比如普通C18在纯水相条件下。相塌陷会导致色谱柱对化合物的保留不足。8 系统压力升高的原因及排除使用者通过观察仪器的系统监测掌握系统的压力,如果发现压力偏高,不要立即认为“柱压高了”,因为系统压力通常由柱前压力、色谱柱压力和流通池压力加合而成。此时正确的做法是:在操作条件下,测定系统压力,得到p总;卸下色谱柱,在相同条件下,测定柱前压力,得到p前;用两通将泵流出管路与流通池连接,在操作条件下,测定压力,得到p(前+后)。通过计算找出压力高是来自柱前、色谱柱还是柱后。http://www.dikma.com.cn/Public/Uploads/images/press%20increase.bmp以上情形假设没有安装保护柱,如果安装了保护柱,压力升高时应先测试保护柱的压力,以便确认问题是否来自保护柱。9 系统压力不稳定通常情况下,泵压的变动值超过了0.5 Mpa,系统压力就属于不稳定的范畴。导致系统压力不稳定的最直接原因为流动相流量和组成输出的不稳定,而导致流动相输出不稳定的原因通常包括溶剂互容性较差、系统漏液、系统存在气泡以及输液系统部件工作失效等等。下面将介绍排除“系统压力不稳”的方法。http://www.dikma.com.cn/Public/Uploads/images/yalibuwen1.bmphttp:
柱色谱法是不是属于固相萃取
近日想配一色谱填充柱,发现有气固和气液两种固定相,不知哪位大哥可告知两者区别呀?
液固色谱的固定相为固体吸附剂,常用硅胶,广泛应用,对具有不同极性取代基的化合物或异构体混合物表现出较高的选择性,对同系物的分离能力较差。液固色谱主要优点是价格便宜,对样品的负载量大,稳定性好,至今仍是[url=https://insevent.instrument.com.cn/t/5p][color=#3333ff]液相色谱[/color][/url]分离中优选方法。液液色谱的固定相是将一种极性或非极性固定液负载在惰性固相载体上,具有再生方便,负载量高,重现性好,分离效果好等优点,但固定液在流动相会产生微量溶解,固定液会不断流失,而流失的固定液又会污染被分离的组分,这使得液液色谱的应用受到了限制。现在化合键合固定相的使用日益广泛,已逐渐取代了液液色谱。
摘 要:综述了高效液相色谱配体交换色谱手性固定相的发展、制备及其在手性拆分中的应用,讨论了洗速、进样量、中心金属离子及其浓度、流动相pH 值、柱温、有机改性剂等对对映体分离的影响,阐述了对手性识别机理的认识.[img]http://www.instrument.com.cn/bbs/images/affix.gif[/img][url=http://www.instrument.com.cn/bbs/download.asp?ID=23983]高效液相色谱手性拆分中的配体交换色谱手性固定相[/url]
声明:本文系chenjunxian的原创,征得陈老师的同意,帮其贴出来参加原创大赛,敬请各位大力支持!PXC固相萃取一高效液相色谱法测定四川泡菜中的多菌灵、克百威摘要:通过对四川泡菜进行固相萃取一高效液相色谱法测定多菌灵、克百威残留量。样品经乙酸乙酯提取后,PXC固相萃取柱提取净化,HPLC法测定。在添加水平为1,5,20μg/g时,多菌灵的回收率在86.3%一92.3%之间;RSD5%(n=6),检出限为0.03mg/kg,克百威的回收率在76.3%一89.4%之间;RSD5% (n=6),检出限为0.06mg/kg,该方法的测定结果满足农药残留量的检测要求。关键词:固相萃取;高效液相色谱法;四川泡菜;多菌灵;克百威多菌灵多菌灵又名棉萎灵、苯并咪唑44号,结构式为http://bbs.instrument.com.cn/xheditor/xheditor_skin/blank.gif。多菌灵是一种广谱性杀菌剂,对多种作物由真菌(如半知菌、多子囊菌)引起的病害有防治效果。可用于叶面喷雾、种子处理和土壤处理等。能防治水稻、棉花、四川泡菜、果树等多种作物的多种病害,但它的残效期比较长,对哺乳动物有一定的毒性,因此农产品中多菌灵残留量的测定越来越受到重视。克百威化学名称为:2,3一二氮-2,2一二甲基苯并吠喃-7一基N一甲基氨基甲酸醋,又名呋喃丹,结构式为:http://bbs.instrument.com.cn/xheditor/xheditor_skin/blank.gif,是一种广谱性杀虫、杀线虫剂,具有触杀和胃毒作用。它与胆碱酯酶结合不可逆,因此毒性甚高。能被植物根部吸收,并输送到植物各器官,以叶缘最多。土壤处量残效期长,稻田水面撒施残效期短。适用于水稻、棉花、烟草、大豆等作物上多种害虫的防治,也可专门用作种子处理剂使用。克百威毒性高,使用时只批准拌种用,严禁喷施克百威,但由于克百威杀虫效果好,被个别不法人员用在蔬菜的产生加工环节上,由于四川泡菜具有生产集中、流通广等特点,如出现食品安全事件涉及范围广,因此要求快速、灵敏、准确地同时检测出未知农药残留,对检测技术提出了新的要求。在食品安全监管工作中除做好宣传工作外,对其
固相微萃取是20世纪80年代末才出现的样品分析前处理技术,其具有选择性好,样品用量少, 无溶剂、操作简单、快速、费用低,并与气相色谱或液相色谱直接联用等优点。目前,固相微萃取-气相色谱/质谱在环境分析、药物分析、食品分析等领域的应用得到了广泛应用,尤其在水果品种快速筛选、鲜花头香成分的活体分析、天然调味料或中草药的快速真假鉴别上表现出更好的发展势头。小茴香为伞形科植物茴香的干燥成熟果实, 是常用的辛香调味料,也是药典收录的一味中药, 具有散寒止痛, 理气和胃的功能,可用于寒疝腹痛, 脘腹胀痛,食少吐泻等。小茴香的混淆品主要有时萝子、葛缕子等。已有的关于小茴香挥发性成分报道,多数是对小茴香水蒸气蒸馏挥发油进行分析得出的结果。水蒸气蒸馏方法,设备简单,但操作时间长,提取过程中,有些敏感性成分会发生化学变化,或引入外来干扰成分。本文采用SPME/GC-MS对小茴香挥发性成分进行研究,为建立小茴香快速分析鉴定方法,全面了解小茴香的挥发性成分,进一步开发利用我国的小茴香资源提供基础。
[color=#444444]我做标准曲线,用的单组分进样可总是在出现各种单组分峰后,固定时间时都有一个峰(所有色谱条件一致)[/color]

作者:http://d.g.wanfangdata.com.cn.www.auth.njfu.edu.cn/Images/head_pic.gif陈翊鲲学科专业:环境科学授予学位:硕士学位授予单位:华南师范大学导师姓名:卢平本文研究采用多壁碳纳米管作为固相萃取固定相吸附剂,并自制成固相萃取柱,建立离线固相萃取的预处理方法对环境样品包括食品、河水等进行前处理净化分离,采用高效液相色谱法作为检测手段,对环境样品中的丙烯酰胺含量进行分析检测。应用该方法对超市中出售的虾条和薯片样品、南洲水厂以及珠江官洲河段河水等环境样品进行了检测。 本文采用购自深圳市纳米港有限公司已纯化的多壁碳纳米管作为固相萃取的吸附剂对样品进行预处理,并从六种不同规格的多壁碳纳米管中确定了规格孔径为40~60nm,长度为1~2μm的多壁碳纳米管对丙烯酰胺有较好的吸附效果,并使用该规格的多壁碳纳米管作为固相萃取吸附剂,结合高效液相色谱法,采用紫外检测器在室温条件下使用Diamonsil 5u C18色谱柱进行色谱分离,10%的甲醇水溶液作为流动相,对环境样品中丙烯酰胺含量进行了检测。 建立了以多壁碳纳米管作为固相萃取固定相吸附剂,结合高效液相色谱作为检测手段,对环境样品中丙烯酰胺含量的检测方法。该方法的结果显示,经多壁碳纳米管处理后各种待测样品的加标回收率在87.19%~92.28%之间,相对标准偏差为2.51%,检出限为5 μg/L。可见该方法符合实际检测分析的要求。 本文还针对多壁碳纳米管的吸附净化性能使用活性炭颗粒进行对比实验,建立了以活性炭颗粒作为固相萃取固定相吸附剂,结合高效液相色谱作为检测手段,对食品中虾条样品的丙烯酰胺含量的检测方法。实验结果显示经活性炭颗粒处理后虾条测样品的加标回收率在77.8%~84.3%之间,相对标准偏差为3.90%。http://ng1.17img.cn/bbsfiles/images/2012/08/201208271728_386587_2379123_3.jpg







 我要推广仪器
我要推广仪器
 下载APP
下载APP