刚刚开始做[url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url],目前一头雾水,使用的是国产的仪器,测定有机溶剂中醇类、苯类的含量,使用的是内标法计算,但是最终结果是含量确不正确,例如我10ml的溶剂中检测的醇类的质量加起来都超过20g,或者算质量百分含量单个组分结果都超过了100%,想请教一下关于好的计算方法,一定要用内标吗?
色谱类方法验证时,方法给出的检出限高于标准判定值,这个方法的检出限要怎么验证呢?我们色谱类方法验证采用信噪比方式验证的不是标准偏差。
[color=#444444]最近收到一份液相样品,样品里有大量的水,有机成分主要是一元和二元醇,醇的碳原子大概在10~15个,样品里可能还含有少量的有机酸和硫酸。现在拿到测试中心想做液相色谱或者液、质联用,目的是了解样品中的醇类都有哪几种,分别占到什么样的比例,但是测试中心要求提供相应的测试方法。请问根据已知内容能否确定液相色谱、质谱的具体设置方法?应该如何设置?[/color]
请问各位同行,巯基类药品该怎么分析含量(找不到基准)化学方法,[url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]和液相色谱的分析方法都可以!
多酚类物质使用质谱法测定的较多,这主要是避免了杂质干扰,但从仪器配备的广泛性和准确定量的角度,[url=https://insevent.instrument.com.cn/t/5p][color=#3333ff]液相色谱[/color][/url]法是比较容易接受的方法。文献报道了利用Hypersil Gold柱,流动相为1%乙酸和甲醇,梯度洗脱,1针70min,流速0.8 ml/min,进样量5微升,建立了没食子酸、新绿原酸、(+)-儿茶素水合物、绿原酸、香草酸、咖啡酸、丁香酸、(-)-表儿茶素、对香豆酸、阿魏酸、芥子酸酸、水杨酸、鞣花酸、芦丁、杨梅素、槲皮素和山奈酚16种多酚类物质的[url=https://insevent.instrument.com.cn/t/5p][color=#3333ff]液相色谱[/color][/url]方法。在仪器配备和方法上可操作性强,能为相关研究者提供有益借鉴。详见[url]https://doi.org/10.3390/horticulturae8020084[/url]
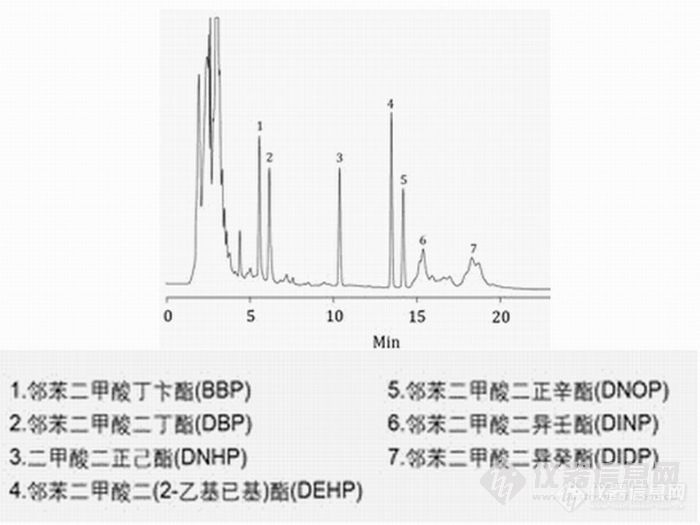
塑化剂邻苯二甲酸酯类的液相色谱方法应用摘要 最近,台湾在食品添加剂中违法加入有害健康的塑化剂邻苯二甲酸酯类物质,导致多家知名饮料及食品污染,这其中包括运动饮料、果汁、茶饮料、果酱、果冻、胶锭粉类产品和食品添加剂等。随着邻苯二甲酸酯类的大量使用,对食品安全和生态环境构成了严重的威胁,因此国内外已将此类化合物列为优先控制污染物。南京科捷针对这一事件,研究了邻苯二甲酸酯类的HPLC检测方法。 对于食品中邻苯二甲酸酯的检测,主要参照GB/T21911-2008《食品中邻苯二甲酸酯的测定》,另外由于大部分客户没有配备GC/MS,可以考虑用液相色谱法进行检测,从而解决食品添加剂的检测问题。关键词 食品饮料添加剂分析 塑化剂 邻苯二甲酸酯分析 邻苯二甲酸酯检测 邻苯二甲酸酯 液相色谱 一.塑化剂 ( 邻苯二甲酸酯)液相色谱图http://ng1.17img.cn/bbsfiles/images/2011/06/201106101652_299157_2242538_3.jpg二.方法引用标准及适用范围本方法适用于食品中16种邻苯二甲酸酯类物质。含油脂样品中各邻苯二甲酸酯化合物的检出限为1.5mg/kg,不含油脂样品中各邻苯二甲酸酯化合物的检出限为0.05mg/kg。采用国家标准GB/T21911-2008进行检测,检测产品种类可涉及饮料、果汁、果冻、食品添加剂配制品等化妆品、儿童玩具、食品包装中,如果其含量超标,会对人体健康产生很大危害。要使用方法国标GB/T21911-2008《食品中邻苯二甲酸酯的测定》。三.16种邻苯二甲酸类物质成分邻苯二甲酸二甲酯(DMP)、邻苯二甲酸二乙酯(DEP)、邻苯二甲酸二异丁酯(DIBP)、邻苯二甲酸二丁酯 (DBP)、邻苯二甲酸二(2-甲氧基)乙酯(DMEP)、邻苯二甲酸二(4-甲基-2-戊基)酯(BMPP)、邻苯二甲酸二(2-乙氧基)乙酯 (DEEP)、邻苯二甲酸二戊酯(DPP)、邻苯二甲酸二己酯(DHXP)、邻苯二甲酸丁基苄基酯(BBP)、邻苯二甲酸二(2-丁氧基)乙酯 (DBEP)、邻苯二甲酸二环己酯(DCHP)、邻苯二甲酸二(2-乙基)己酯(DEHP)、邻苯二甲酸二苯酯、邻苯二甲酸二正辛酯(DNOP)、邻苯二 甲酸二壬酯(DNP)四.液相色谱仪器配置(技术咨询025-84372573)检测项目食品中邻苯二甲酸酯的测定本项目实验单位南京科捷分析仪器应用研究所仪器型号及配置LC-600液相色谱仪P600宝石恒流泵 2台UV600紫外检测器 1台进口原装7725i进样阀 1只WS600色谱工作站 1套混合器 1套液相色谱柱
HJ/T68-2001方法测固定污染源苯胺类,用什么色谱柱?标准上是玻璃填充柱,可以用毛细管柱吗还有采样用的硅胶吸附管都是自己自制的吗?有卖的成品吗?
请问用[url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]方法分析酞酸酯类物质,选用什么样的柱子比较好,谢谢!!
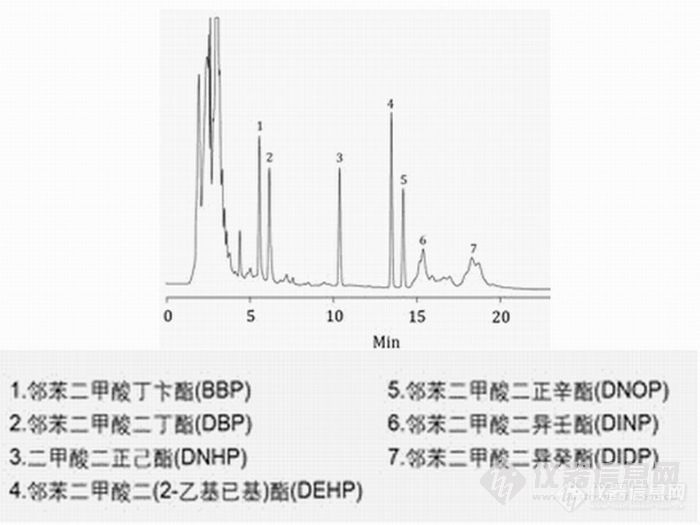
塑化剂邻苯二甲酸酯类的液相色谱方法应用摘要 最近,台湾在食品添加剂中违法加入有害健康的塑化剂邻苯二甲酸酯类物质,导致多家知名饮料及食品污染,这其中包括运动饮料、果汁、茶饮料、果酱、果冻、胶锭粉类产品和食品添加剂等。随着邻苯二甲酸酯类的大量使用,对食品安全和生态环境构成了严重的威胁,因此国内外已将此类化合物列为优先控制污染物。南京科捷针对这一事件,研究了邻苯二甲酸酯类的HPLC检测方法。 对于食品中邻苯二甲酸酯的检测,主要参照GB/T21911-2008《食品中邻苯二甲酸酯的测定》,另外由于大部分客户没有配备GC/MS,可以考虑用液相色谱法进行检测,从而解决食品添加剂的检测问题。关键词 食品饮料添加剂分析 塑化剂 邻苯二甲酸酯分析 邻苯二甲酸酯检测 邻苯二甲酸酯 液相色谱 一.塑化剂 ( 邻苯二甲酸酯)液相色谱图http://ng1.17img.cn/bbsfiles/images/2011/06/201106111518_299244_1641058_3.jpg二.方法引用标准及适用范围本方法适用于食品中16种邻苯二甲酸酯类物质。含油脂样品中各邻苯二甲酸酯化合物的检出限为1.5mg/kg,不含油脂样品中各邻苯二甲酸酯化合物的检出限为0.05mg/kg。采用国家标准GB/T21911-2008进行检测,检测产品种类可涉及饮料、果汁、果冻、食品添加剂配制品等化妆品、儿童玩具、食品包装中,如果其含量超标,会对人体健康产生很大危害。要使用方法国标GB/T21911-2008《食品中邻苯二甲酸酯的测定》。三.16种邻苯二甲酸类物质成分邻苯二甲酸二甲酯(DMP)、邻苯二甲酸二乙酯(DEP)、邻苯二甲酸二异丁酯(DIBP)、邻苯二甲酸二丁酯 (DBP)、邻苯二甲酸二(2-甲氧基)乙酯(DMEP)、邻苯二甲酸二(4-甲基-2-戊基)酯(BMPP)、邻苯二甲酸二(2-乙氧基)乙酯 (DEEP)、邻苯二甲酸二戊酯(DPP)、邻苯二甲酸二己酯(DHXP)、邻苯二甲酸丁基苄基酯(BBP)、邻苯二甲酸二(2-丁氧基)乙酯 (DBEP)、邻苯二甲酸二环己酯(DCHP)、邻苯二甲酸二(2-乙基)己酯(DEHP)、邻苯二甲酸二苯酯、邻苯二甲酸二正辛酯(DNOP)、邻苯二 甲酸二壬酯(DNP)四.液相色谱仪器配置 检测项目 食品中邻苯二甲酸酯的测定本项目实验单位 南京科捷分析仪器应用研究所仪器型号及配置 LC-600液相色谱仪P600宝石恒流泵 2台UV600紫外检测器 1台进口原装7725i进样阀 1只WS600色谱工作站 1套混合器 1套液相色谱柱 进口C184.6mm*250mm*5um色谱柱其它配置 邻苯二甲酸酯标准品PEP玻璃管SPE前处理小柱等来源:http://bbs.antpedia.com/thread-63498-1-1.html
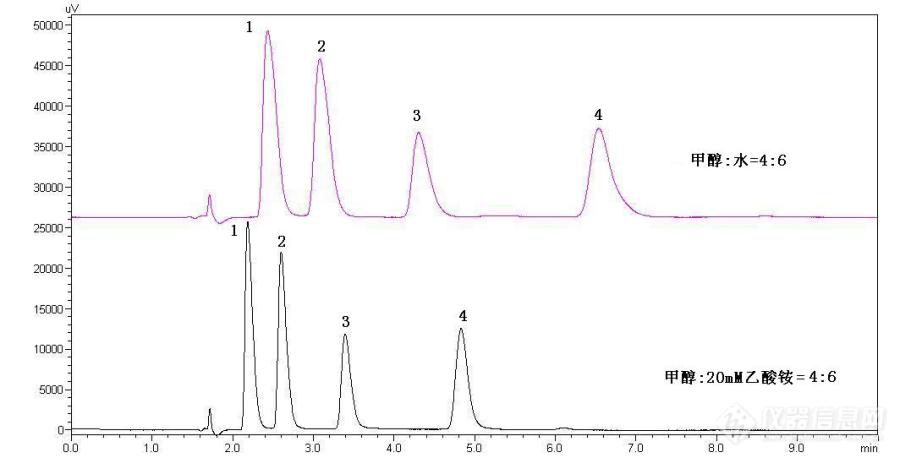
食品中对羟基苯甲酸酯类液相与[url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]法的测定方法比较摘要:本文详细讲述了液相色谱与[url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]法测定食品中对羟基苯甲酸酯类各自的优缺点,GB 5009.31-2016只收录了[url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]法测定标准的测定方法。关键词:对羟基苯甲酸酯类;液相色谱;[url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url];优缺点;GB 5009.31-2016引言[font=黑体][size=18px][b] [/b][/size][/font][font=宋体][size=12px]食品中常见的对羟基苯甲酸酯类又称为对羟基安息香酸酯或尼泊尔金酯,包括对羟基苯甲酸甲酯、乙酯、丙酯、丁酯,是一类新一代高效低毒消毒杀菌防腐剂,它的抗菌能力、pH应用范围及用量比苯甲酸和山梨酸及其盐类广(见表1),且使用安全,经济方便,对人体刺激较小。在国外,已被广泛用于食品、饮料、化妆品和医药等方面。作为食品防腐剂,它可用于饮料、果蔬加工品、海产加工品、禽畜加工品、调味品、啤酒、米酒等加工品中,还可用于水果、蔬菜和海产品的防腐保鲜。它不但可完全替代苯甲酸钠和山梨酸钾,其使用范围比苯甲酸钠和山梨酸钾更广。在国外,已被广泛用于食品、饮料、化妆品和医药等方面。在日本,对羟基苯甲酸酯和山梨酸是主要的防腐剂产品。而我国,对羟基苯甲酸酯类防腐剂的用量也在逐年增加,成为防腐剂的第二个主要产品。[/size][/font][align=center]表1 GB2760-2014对羟基苯甲酸酯类限量要求[/align][img]https://ng1.17img.cn/bbsfiles/images/2020/08/202008192125459013_3455_2166779_3.png[/img]实验部分讨论液相色谱1.前处理及色谱分析条件称取5g(精确至0.01g)试样于50ml比色管中,加入15mL95%乙醇,混匀,超声波清洗器提取10min,冷却至室温后用95%乙醇定容至50mL刻度线,摇匀。静置分层,取上清液经0.22um微孔滤膜过滤后待测。色谱柱:C18柱,150×4.6 mm(i.d),5 μm,或性能相当者;流动相:甲醇(B)+20 mmol/L乙酸铵溶液(A) = 40+60(体积比);洗脱梯度见表2:[font=黑体]表2. 对羟基苯甲酸酯类的洗脱程序[/font][table][tr][td]时间/min[/td][td]2.00[/td][td]4.00[/td][td]12.00[/td][td]12.01[/td][td]15.00[/td][/tr][tr][td]B%[/td][td]40[/td][td]60[/td][td]60[/td][td]40[/td][td]stop[/td][/tr][/table]流速:1 mL/min;柱温:35 ℃;进样体积:10 μL;检测波长扫描范围:210 nm—390 nm,定量波长256 nm。由于对羟基苯甲酸酯在[font=times new roman]pH4~8的[/font]范围内稳定存在且有很好的抗菌效果,但水溶性较低,易溶于乙醇,而乙醇的毒性较甲醇低,所以采用乙醇做为标准使用液和样品提取液。大部分添加对羟基苯甲酸酯类的食品都具有含水性高、不易长期保存的特点,因此在取样时选择被测物稳定保存的状态,采用浸泡过夜、超声及振荡提取的方式,能达到较好的提取效果。2、色谱条件的选择及优化2.1 检测波长的选择以浓度为[font=times new roman]0.02 mg/ml的标准使用液依次在高效液相色谱—二极管阵列检测器190 nm-410 nm波长[/font]范围进行扫描,以确定被[font=times new roman]测物质的最大吸收波长,对羟基苯甲酸酯类的最大吸收波长均十分相似,在256nm处有紫外最大吸收峰,这是与自身具有苯环和羰基结构所决定,实验选择256nm为检测波长,能有效满足四种物质的分析检测。[/font] [size=12px] 2.2 分析条件的选择[/size]分别使用水和20mM乙酸铵做流动相在相同色谱条件下进行分析(见图2),UPLC(超高速液相)在5分钟内完全分离且重现性好,出峰顺序依次为对羟基苯甲酸甲酯、乙酯、丙酯、丁酯。由图可以看出在峰形和分析时间上利用缓冲盐做流动相要优于纯水,特别是面对食品样品的复杂性,选择20mM乙酸铵和甲醇作为流动相能有效排除基质干扰。[img]https://ng1.17img.cn/bbsfiles/images/2020/08/202008192125462304_3941_2166779_3.jpg[/img]图2 混合标准样品色谱图1.对羟基苯甲酸甲酯[font=arial][color=black];[/color][/font][font=times new roman]2.[/font]对羟基苯甲酸乙酯[font=arial][color=black];[/color][/font][font=times new roman]3.[/font]对羟基苯甲酸丙酯[font=arial][color=black];[/color][/font][font=times new roman]4.[/font]对羟基苯甲酸丁酯实验选择C18色谱柱进行分离,是根据尼泊尔金酯类的物理化学性质,在C18柱上具有良好的保留,当提高有机相甲醇的比例时,能快速得到洗脱。选择20m M乙酸铵作为流动相,能较好的平衡食品复杂基质的p H值,有效避免杂质干扰。实验尝试使用了日本岛津HPLC-20A,美国Grance Alltima 4.6mm×150mm C18色谱柱与日本岛津UPLC,shim-pack XR-ODS 3.0mm×75mm C18色谱柱对市售果蔬汁饮料、酱腌菜及酱油制品和糕点等所含的对羟基苯甲酸酯类进行比对分析,发现均能得到良好分离,且结果一致。在实验中发现,相同条件下,采用HPLC-20A,美国Grance Alltima 4.6mm×150mm C18色谱柱对酱腌菜类食品进行分析,在对羟基苯甲酸乙酯处会有干扰(见图3),通常情况下,改变流动相的比例能避免此类问题的发生,但若直接选用岛津UPLC,shim-pack XR-ODS 3.0 mm×75 mm C18色谱柱进行分析,则能有效避免(见图4)。考虑到两种C18柱的价格及维护费用,选择使用Grance Alltima 4.6mm×150mm C18进行大批量的实验分析,在保证实验数据的准确可靠前提下,能有效降低成本。[img]https://ng1.17img.cn/bbsfiles/images/2020/08/202008192125466015_1144_2166779_3.jpg[/img] [img]https://ng1.17img.cn/bbsfiles/images/2020/08/202008192125467275_3703_2166779_3.jpg[/img]图3 酱腌菜中对羟基苯甲酸酯类色谱图(HPLC-20A)峰序同图2 图4 酱腌菜中对羟基苯甲酸酯类色谱图(UPLC)2.3 基质的干扰与条件优化 [size=16px] [/size][size=12px]实验选择糕点、果蔬汁饮料及酱油及酱腌菜类为基质,采用上述方法进行前处理,岛津HPLC-20A,Grance Alltima 4.6mm×150mm C18进行分析检测,发现果蔬汁及饮料等,基质简单,峰形对称(见图5);在进行酿造酱油基质的加标回收分析时发现,甲酯由于出峰时间较早,容易被杂质峰包埋,致使检测的检出限降低(见图6);酱腌菜类食品由于基质复杂,特别是在腌制过程中产生的不明物质较多且各有差异,在实验中稍有不慎,极容易与对羟基苯甲酸乙酯、丙酯产生干扰(见图7),因此在进行分析过程中应注意色谱条件的选择,改变流动相中甲醇的比例能有效避免杂质的干扰。[/size][img=,690,331]https://ng1.17img.cn/bbsfiles/images/2020/08/202008192131109762_6420_2166779_3.png!w690x331.jpg[/img][img=,690,308]https://ng1.17img.cn/bbsfiles/images/2020/08/202008192131386200_7631_2166779_3.png!w690x308.jpg[/img][img=,690,325]https://ng1.17img.cn/bbsfiles/images/2020/08/202008192131527267_1909_2166779_3.png!w690x325.jpg[/img][url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]国标GB5009.31-2016《食品安全国家标准 对羟基苯甲酸酯类的测定》检测范围为酱油、醋、饮料及果酱,采用GC酸化提取后进行分析检测,实验原理与本方法可互为验证和补充,但操作步骤复杂,耗时长,乙醚试剂消耗大,对检测人员伤害较大,且效率低,但基质处理的干净,不存在干扰而产生假阳性的情况。仪器条件:Agilent 7890 检测器:FID 进样量:1 μL 色谱柱:[color=black]HP-1(30m*320μm*0.25μm);HP-5(30m*320μm*0.25μm) ;DB-17 30m*320μm*0.25μm ; [/color][color=red]HP-5MS(30m*320μm*0.25μm)[/color][color=red] [/color]检测器温度:240[font=宋体]℃[/font] 进样温度:250[font=宋体]℃[/font]程序升温:100[font=宋体]℃[/font](1min) 20 [font=宋体]℃[/font]/min 160[font=宋体]℃[/font](3min) 15 [font=宋体]℃[/font]/min 250[font=宋体]℃[/font](3min)样品处理:取样5.0 g(±0.01 g)于50 mL塑料离心管中,加入1 mL盐酸(1:1)溶液酸化,再加入10 mL饱和氯化钠水溶液摇匀,分别每次30 mL乙醚提取三次,涡旋振荡4000 r/min离心,取上清液乙醚合并入250 mL分液漏斗中,先加入10 mL饱和氯化钠水溶液洗涤一次弃去水层,分别每次30 mL 1g/100mL的碳酸氢钠溶液洗涤洗三次 ,静置,弃去水层。过装有10 g无水硫酸钠的漏斗至鸡心瓶中,35[font=宋体]℃[/font]浓缩至干,用无水乙醇定容至2 mL上机测试。注释:[color=red](1)使用HP-5MS色谱柱主要是为了增加分离度,使得目标峰与分析纯乙醚中的干扰峰分离开。[/color](2)对羟基苯甲酸乙酯处有试剂干扰,来自乙醚。(3) 标准中使用125 mL的分液漏斗,实验室直接使用50mL的塑料离心管更易于提取。(4) 标准中用75 mL、50 mL、50 mL乙醚提取三次,实验室用离心管离心取上层乙醚层更方便,由于离心管容积只有50 mL,所以减少乙醚量,分别每次用20 mL乙醚提取三次。(5) 标准中用分液漏斗萃取,静置弃去水层,实验室用离心的方法分层,取上清液乙醚层。(6) 标准中在分液漏斗中加10 g无水硫酸钠于室温放置30 min脱水,实验室过装有10 g无水硫酸钠的漏斗脱水。色谱图分析比较:通过图8、图9([url=https://insevent.instrument.com.cn/t/Mp]气相[/url]法前处理)与图5~图7(液相法前处理)比较,[url=https://insevent.instrument.com.cn/t/Mp]气相[/url]的图谱明显干净多了,而且不存在一点的基质干扰的现象。 [img]https://ng1.17img.cn/bbsfiles/images/2020/08/202008192125470058_269_2166779_3.png[/img] [img]https://ng1.17img.cn/bbsfiles/images/2020/08/202008192125470897_3417_2166779_3.png[/img]果酱食品基质加标(2ppm) b. 果酱类食品空白基质图8 果酱食品中对羟基苯甲酸酯类空白基质及基质加标(2ppm) [img]https://ng1.17img.cn/bbsfiles/images/2020/08/202008192125471786_7777_2166779_3.png[/img] [img]https://ng1.17img.cn/bbsfiles/images/2020/08/202008192125472549_3883_2166779_3.png[/img] a. [size=12px]果酱食品基质加标(2ppm) b. 果酱食品空白基质 [/size] [size=12px]图9 果酱食品中对羟基苯甲酸酯类空白基质及基质加标(2ppm) [/size]按国标GB5009.31-2016检测方法线性范围和测定低限:在1~500 μg/mL浓度范围内,以峰面积(y)与目标化合物浓度(x,μg/mL)绘制标准工作曲线。结果表明,在1.0~500 μg/mL质量浓度范围内,目标化合物良好线性关系,线性方程式、线性关系和测定低限(LOQ,S/N=10)见表2。表2 线性方程式、线性相关系数和定量限[table][tr][td][align=center]化合物[/align][/td][td][align=center]线性方程式[/align][/td][td][align=center]线性相关系数[/align][/td][td][align=center]定量限/(mg/kg)[/align][/td][/tr][tr][td][align=center]对羟基苯甲酸甲酯[/align][/td][td][align=center]Y=0.532847x-1.26878[/align][/td][td][align=center]0.99915[/align][/td][td][align=center]2.0[/align][/td][/tr][tr][td][align=center]对羟基苯甲酸乙酯[/align][/td][td][align=center]Y=0.563352x-1.13532[/align][/td][td][align=center]0.99972[/align][/td][td][align=center]2.0[/align][/td][/tr][tr][td][align=center]对羟基苯甲酸丙酯[/align][/td][td][align=center]Y=0.554134x-2.50160[/align][/td][td][align=center]0.99915[/align][/td][td][align=center]2.0[/align][/td][/tr][tr][td][align=center]对羟基苯甲酸丁酯[/align][/td][td][align=center]Y=0.530932x-4.29406[/align][/td][td][align=center]0.99971[/align][/td][td][align=center]2.0[/align][/td][/tr][/table] 回收率和精密度分别向空白蚝油中添加目标物,做空白添加回收试验,添加水平为2.0、5.0、50.0 mg/kg,各添加水平分别做6次平行试验。加标回收率为88.7%~108%,相对标准偏差(RSD)为1.3~5.2%,方法的精密度及回收率均满足定量测定的要求,试验结果见表3。[size=12px][font=times new roman] [img=,690,371]https://ng1.17img.cn/bbsfiles/images/2020/08/202008192133361979_6058_2166779_3.png!w690x371.jpg[/img][/font][/size] 总结: [font=宋体][size=12px]国标方法是用盐酸酸化样品,乙醚提取,浓缩后,用具有氢火焰离子化检测器的[url=https://insevent.instrument.com.cn/t/Mp]气相色谱仪[/url]进行分离检测,外标法定量;耗时长、成本高、消耗大、毒性强、工作效率低等弱点,极大的限制了在食品检测过程中的广泛运用,优点是谱图干净,几乎没有基质干扰的现象。针对食品检测工作中样品量大、基质干扰多、成分复杂且易变质腐败等特点,在检测过程中也需要建立一种准确高效的检测方法运用于实际工作;液相法是一种快捷、准确、适用范围广的方法,以满足检测工作的需要,达到提高工作效率的目的。遇到有基质干扰,不合格的样品时改用[url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]法进行准确定量检测。[/size][/font] [size=12px] [/size]
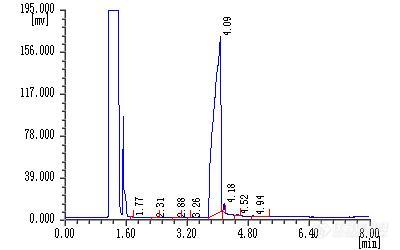
[color=#DC143C]所在地区:陕西西安,从事行业:药物研发分析,分析的物质名称:某胺类物质(涉密,具体名字省略)[/color][color=#00008B]方法名称:胺类盐酸盐[url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]分析方法(自己研发) 企业标准[/color]前些日子,接到一个新项目,要对一个胺类盐酸盐进行纯度分析,该样品为科研新研发的项目,以前从未做过类似的工作,对我来说也是开天辟地了,觉得有点难度。首先拿到反应工艺,了解该物质的化学性质如下:盐酸盐,无紫外吸收,极性大,反相不保留 加上客户更看重GC结果,经过严重的思想斗争后认为做[url=https://insevent.instrument.com.cn/t/Mp]气相[/url]比液相容易点(盐类物质在反相上不保留),着手准备!由于是做纯度,因此面积归一化法足够了,要求不高,无需校正,再说了,新研发的样品中未知杂质太多了,也无法一一校正!1.样品预处理 盐酸盐由于难以汽化无法直接进样分析,需要前处理,必须把盐酸破坏掉,把有机物提取出来,进样分析,相关的文献报道不是很多,自己摸索吧,做成了,皆大欢喜;失败了,也不是很丢人,毕竟对我来说是个尝试,大致思路出来了,做好实验前的准备工作很重要!1.1预处理过程参数的简单选择 (1) 用什么来破坏盐酸盐呢?碱!回答的很正确!备用选项:NaOH水溶液、NaOH的甲醇溶液、甲醇钠、氨水,考虑到水可能会对柱子的固定液有伤害,排除掉含水的三种,分别用NaOH的甲醇溶液、甲醇钠对样品进行处理,两者的破坏效果对分析结果的影响无明显差异,但是后者配制起来危险性较高(不知道有没有用过甲醇钠,用金属钠加到甲醇中,金属钠属于危险品,尽量少用),因此采用NaOH的甲醇溶液作为处理的溶液;由于NaOH在甲醇中的溶解度有限,因此选用的浓度是2%(m/m)。 (2)pH值对分析结果的影响 pH值过小的话,样品可能破坏的不完全,并且样品在低pH值时的稳定性也较差(相对高pH而言),pH值过高担心柱子耐受不了,经过一番实验发现pH值在9时分析结果较让人满意。 (3)样品破坏后的稳定性实验 样品从盐酸盐破坏为胺后在空气中的稳定性较差(容易被氧化,后来经过GC-MS确认是和空气中的氧气反应了),很难保证数据的精密度,因此预处理过程中要对样品进行通N2保护,使数据的精密度得到了保证。 样品经过破坏后,用0.45um滤膜滤掉盐类及部分不溶物,进样分析。2.色谱条件的选择 手头的仪器就是14C,2010,2014C了,这些都不重要,最重要的是选择柱子,能选的柱子包括DB-1,DB-17,DB-FFAP,Rtx-5Amine(新买的,专门做胺类物质的,5000多块大洋哦,蛮心疼的),检测器只有FID,没有优化的空间了;能进行选择的只有柱子了,FFAP一般是用来做挥发性脂肪酸的,这个是胺,大方向都不对,直接排除;DB-17柱子只有两根,怕弄坏了领导怪罪,排除;DB-1超多,随便找一个将要淘汰的先试试(由于样品有碱性,怕对柱子不好,不敢先用好柱子做),Rtx-5Amine胺基柱专门做胺类物质的,可以和DB-1做个对比,看哪个效果好,对比结果如图所示,胺基柱的优势是相当的明显,DB-1排除。经过简单优化后的色谱条件:仪 器:GC-2010分析柱:Rtx-5 Amine(30m*0.32mm*1um)载 气:He检测器:FID分流进样(SPL=30)温度参数设置:INJ 280度,DET 300度,COL 100度起,10度/min升温至280度,停留20min;DB-1做的结果:[img]http://ng1.17img.cn/bbsfiles/images/2009/06/200906111455_155052_1621482_3.jpg[/img]Rtx-5Amine做的结果:[img]http://ng1.17img.cn/bbsfiles/images/2009/06/200906111456_155053_1621482_3.jpg[/img]用到的装备:[img]http://ng1.17img.cn/bbsfiles/images/2009/06/200906111456_155054_1621482_3.jpg[/img]3.0小结 于如期完成了,说不上圆满,因为有些地方还不完善,比如预处理过程有点繁琐,稳定性也有优化的空间等。这个项目结束后,铺天盖地的胺类物质都送过来做GC,超级郁闷!道路还很漫长................当局者迷,旁观者清,请各位专家、版友积极指出方法中的不足和错误地方,作为当事人,不容易发现错误.................
因为GB5750-2006上不是用这个方法检测,各位用FFAP色谱柱测硝基苯类的同仁:请问你们都是用的什么色谱仪,色谱条件是什么,可以分离哪些物质,检测结果怎样(回收率,偏差、检出限)等等?请赐教!谢谢!
购买标准物质时,能不能提供物质色谱图或方法呀?
DB33/T 563-2005 水产品中磺胺类残留量的测定方法 液相色谱法浙江省地方标准。2005-06-30发布,2005-07-15实施,现行有效。[img]http://www.instrument.com.cn/bbs/images/affix.gif[/img][url=http://www.instrument.com.cn/bbs/download.asp?ID=139353]DB33/T 563-2005 水产品中磺胺类残留量的测定方法 液相色谱法[/url]
安捷伦液相色谱-柱后衍生测氨基甲酸酯类农药,用的是NY/T761方法,有做过的同行么。流动相的设置如何优化
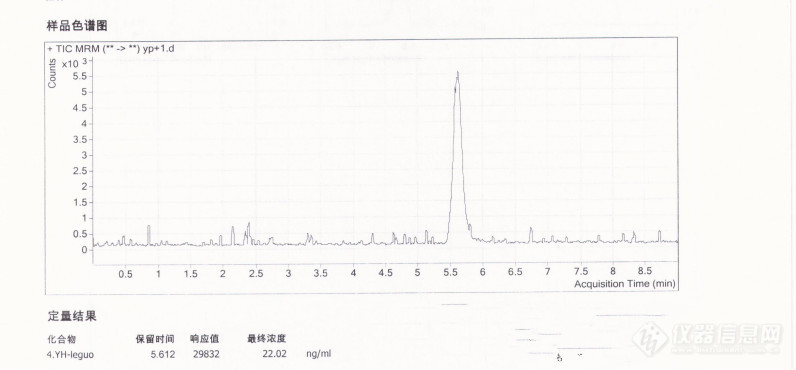
[url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]法与液相色谱质谱法测豆类蔬菜中的氧乐果方法比较[url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]检测豆类(白豆子)中的氧化乐果仪器:岛津[url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]GC2010plus,FPD检测器,程度升温:100度保持1.0min,20度/min 升到230度(保持1min);再以20度/min 升到260度(保持2min);再以40度/min 升到280度(保持1.5min) 色谱柱:RTX-1701柱;进样口:250度,检测器:300度,不分流进样,柱流量:1.0mL/min,样品前处理:1、提取:准确称取25.0克试样于匀浆机中,加入乙腈50.0mL,在匀浆机中高速匀浆2min后过滤,滤液加入7克氯化钠,振荡静置,使乙腈层与水层分层。 2、净化:取10.0mL乙腈提取液于40度水浴旋转蒸发至近干,用丙酮定容至5.0mL,用0.2um滤膜过滤,滤液上[url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]测定,同时进行试剂的空白试验。 3、加标回收(加标原则:如果样品为未检出,按定量限来加标,如果样品为阳性,则加标量约为检出量的3倍):吸取1000ng/mL氧乐果标液2.50mL于25.0克的试样中,与试样同时同样处理,相当于加标水平为0.10mg/kg,最终理论上机浓度为100ng/mL。氧乐果工作曲线浓度:20、50、100、200、300、500ng/mL[img=,690,224]https://ng1.17img.cn/bbsfiles/images/2018/08/201808132124456926_439_2166779_3.png!w690x224.jpg[/img]50ng/mL氧乐果与试剂空白的堆栈色谱图:[img=,690,423]https://ng1.17img.cn/bbsfiles/images/2018/08/201808132128226444_2349_2166779_3.png!w690x423.jpg[/img]从堆栈图可以看出:试剂空白氧乐果出峰处没有任何的干扰。50ng/mL氧乐果与样品的堆栈色谱图:[img=,690,429]https://ng1.17img.cn/bbsfiles/images/2018/08/201808132132374191_2107_2166779_3.png!w690x429.jpg[/img]由工作曲线得出样品的浓度为40.7ng/mL。50ng/mL氧乐果与样品加标的堆栈色谱图:[img=,690,447]https://ng1.17img.cn/bbsfiles/images/2018/08/201808132135100711_1706_2166779_3.png!w690x447.jpg[/img]由工作曲线得出样品加标后的浓度为92.5ng/mL,由此计算出加标回收率为:(92.5-40.7)*100%/100=52%液相色谱质谱法(复测):[img=,686,288]https://ng1.17img.cn/bbsfiles/images/2018/08/201808132147105941_4549_2166779_3.png!w686x288.jpg[/img]加标回收:1000ng/mL3.0mL于样品中,同时同样处理,最终定容体积V1为2.50mL,相当于理论上机浓度为20ng/mL(样品前处理时,最终定容体积V1为0.5mL)。[url=https://insevent.instrument.com.cn/t/Yp][color=#3333ff]液质[/color][/url]测定用的氧乐果系列工作使用液:[img=,690,204]https://ng1.17img.cn/bbsfiles/images/2018/08/201808132155310274_8706_2166779_3.png!w690x204.jpg[/img][img=,677,431]https://ng1.17img.cn/bbsfiles/images/2018/08/201808132156409256_2949_2166779_3.png!w677x431.jpg[/img]20ng/mL氧乐果MRM图:[img=,690,349]https://ng1.17img.cn/bbsfiles/images/2018/08/201808132201453681_7737_2166779_3.png!w690x349.jpg[/img][img=,676,271]https://ng1.17img.cn/bbsfiles/images/2018/08/201808132201512921_7305_2166779_3.png!w676x271.jpg[/img]样品的测定结果为:氧乐果的含量为:20.56*0.5*30*2*0.001/15.0=0.04mg/kg[img=,690,375]https://ng1.17img.cn/bbsfiles/images/2020/10/202010081648437833_8836_2166779_3.jpg!w690x375.jpg[/img]加标样品后MRM图(样品处理:加标回收:1000ng/mL3.0mL于样品中,同时同样处理,最终定容体积V1为2.50mL,相当于理论上机浓度为20ng/mL(样品前处理时,最终定容体积V1为0.5mL)):[img=,690,320]https://ng1.17img.cn/bbsfiles/images/2020/10/202010081646490017_6074_2166779_3.jpg!w690x320.jpg[/img]加标回收率为(22.02ng/mL-0.04(mg/kg)×15g×1000/30/5)/20(加标液理论上机浓度)=90.1%。结论:1、遇到样品不合格时最好能更换检测方法及检测人员。2、[url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]法与液相色谱质谱法测氧乐果的数据相符合,证明两种检测方法的可行性。3、加标回收的原则:如果样品为未检出,按定量限来加标,如果样品为阳性,则加标量约为检出量的3倍
我准备做苯胺类[url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]-质谱法。我先有的标液是甲醇中的,方法说的是用二氯甲烷定容。我拿我的标液直接用二氯甲烷定容做可以吗。两个试剂互溶吗?
请教各位老师一个问题,[url=https://insevent.instrument.com.cn/t/Mp][color=#3333ff]气相色谱[/color][/url],[url=https://insevent.instrument.com.cn/t/5p][color=#3333ff]液相色谱[/color][/url]和质谱类的仪器如何做校准后的确认,很多方法都要用到该仪器,需要把每个方法都做确认吗?谢谢各位老师了。
安捷伦7890A气相色谱测定甲乙丙丁戊5种醇类,顶空进样,溶剂是乙酸乙酯,使用的柱子是DB-624(30x0.25x0.14),进样口温度180,流量1.2,顶空瓶放入配置好的标准溶液10ul,甲醇峰经常出现平顶峰或者圆顶峰的现象,按照故障排除的方法,不断减小进样量和加大分流比,调试到峰比较正常一点的时候,信号值只有20PA左右,峰面积大概也只有100左右,这样正常吗?正常的时候,其它四个醇类的峰都比较正常,但是甲醇峰始终还是有一点拖尾,这是怎么回事呢?
多糖类手性固定相在色谱中的应用郑 芸 方积年(中国科学院上海生命科学院上海药物研究所,上海201203)摘要 手性色谱技术是最重要的手性分离方法之一,它不仅可以快速地分析对映体纯度,也可以用于大量制备光学异构体。设计和发展高效的固定相是手性色谱技术的核心。在诸多的手性固定相中,多糖类手性固定相因品种繁多、耐用而被广泛应用。本文综述了多糖类手性固定相在高效液相色谱、模拟移动床色谱、超临界流体色谱及膜分离中的应用。共引用文献52篇。关键词 多糖,手性固定相,色谱,评述1 引 言 近20年来,用色谱方法分离手性化合物取得了显著进展,已广泛应用于许多领域,如药物化学、不对称合成和生物分析等,不仅可以测定光学纯度,也可用于大量制备光学异构体。 手性色谱技术的核心是设计和制备适用范围广的手性固定相(chiral stationary phase,CSP)。至今已制备出大量用于色谱的CSP,其中120多种已商品化。CSP可分为两大类:一类是由小分子固定在硅胶载体上构成(刷型或Pirkle型),另一类是用光学聚合物固定在载体上制成,多孔胶状的聚合物也可直接用作CSP。其中Okamoto等发展的多糖类固定相是非常有用的分离工具,它们种类繁多、耐用而且负荷量大。其它广泛使用的手性固定相有衍生化的酒石酸CSP(Kromasil—TBB) ]、a1一酸性糖蛋白、Pirkle固定相、环糊精、聚丙烯酰胺和大环抗生素,如万古霉素、teicoplanin和瑞斯托菌素以及最新的用分子印记技术及仿生传感技术发展的CSP 。 多糖,如纤维素和淀粉是自然界大量存在的有光学活性的生物聚合物。它们具有良好的精细结构,能拆分异构体,包括氨基酸衍生物和联苯衍生物的阻转异构体,但它们的手性识别能力不强,适用面也很窄,只能用于毛细管电泳(CE)分析中。半合成的经过改性的多糖适用范围则大大扩展,可用于LC、CE、SFC、TLC、膜分离及萃取中,既可用于分析也可用于制备。经研究发现,多糖类衍生物的手性识别能力与单糖残基的性质、连接位置和连接形式有关。2 高效液相色谱(HPLC) 多糖类手性固定相在HPLC中的应用相当广泛,常见的商品化多糖类手性固定相及应用实例可参考相关文献。纤维素类多糖为刚性的线形结构,而淀粉类多糖具有螺旋形结构。据报道有84%的小分子外消旋化合物可用Chiralcel OJ、Chiralcel OD、Chiralpak AD、Chiralpak AS分离 。用HPLC分析对映体时,除了常用的UV或示差折光指数检测器,还可使用专门检测手性物质的旋光检测器和圆二色散检测器。这也是HPLC比[url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]和NMR光谱等其它分析方法的优越之处。 为提高手性分离效果以利于检测,还可以对样品进行适当的衍生化。Fukushima 用荧光试剂DBD—PZ([4.[(N,N—dimethylamino)一sulfony1]-7一piperazino-2,1,3-benzoxadiazole])和DBD—COHz(4[[(N—hydrazinoformy1)methy1]一N—methy1]amino-7一[N,N一(dimethylamino)sulfony1]-2,1,3-benzoxadiazole)对(RS)-2一芳基丙酸类化合物进行了衍生化,并发现衍生物洗脱顺序发生改变。3 动态高效液相色谱(DHPLC) 新发展的手性DHPLC方法 可用于研究高温时立体化学稳定的手性化合物,它可得到一系列受温度控制的平顶或峰形曲线,从而可以考察对映体互变的动态过程、动力学数据及对映体互变的能垒。一般在CSP上用色谱方法分离外消旋混合物,最多可以得到收率50% 的两种纯的光学异构体。而在DHPLC中,利用CSP来达到对映体互变平衡,从而使分离和平衡合二为一,理论上可以从外消旋混合物中以100%收率得到一种纯的光学异构体。它的基本原理是外消旋混合物立体化学稳定在较低温度时对映体互变过程被抑制,而较高温度发生对映体互变。实验中让外消旋混合物先通过一个低温CSP柱子,将先洗脱出来的组分(A)继续通人第二个高温CSP柱子,收集后洗脱组分(B)。(A)进入第二个柱子后停留足够长时间达到对映体互变平衡,再继续洗脱,得到(A)和(B)。如果进行多次循环平衡、过柱,则可得到纯的对映体(B)。如Lorenz等用DHPLC分离一螺环化合物,该化合物可通过螺环处的C—O 键开环和闭环进行对映体互变。将它依次通过0℃和40℃ 的两根Chiralcel OD柱,平衡2h,即可得到32% ee(enantiomerie excess,ee)的(+)一对映体。4 模拟移动床色谱(SMB) 至今批次处理色谱在应用中仍占主导地位,但大规模制备需要大量CSP。CSP价格昂贵,而且产品的浓度低,洗脱液消耗量大,难以回收。SMB可以节省90% 的流动相并得到更高的产率。在批次处理色谱中被分离组分在流动相的驱动力下移动,固定相只有一小部分起作用。在移动床色谱中,不仅流动相发生移动,固定相也要向相反方向移动,易洗脱的化合物(萃余液)随流动相移动,难洗脱的化合物(萃取液)随固定相移动。整个固定相的分离能力被持续利用,明显地提高了系统产率。但就技术而言很难移动固定相,因此采用模拟方式,SMB的环状柱子实际上是用许多小柱依次连接而成,有规律地改变进样口和出样口,可以达到和固定相移动相同的效果。SMB技术起于20世纪60年代UOP(Universal Oil Products,Des Plaines,IL,USA)从C8 中分离对二甲苯,后来该技术被广泛用于制药工业,以获得光学纯药物。其中应用于SMB的CSP约有70%是多糖类CSP。如Nagamatsu等用SMB方法替代以前的非对映体结晶的方法,用稍做改性的Chiralcel OF(cellulose 4-chlorophenyl carbamate)分离了一种制药工业的中间体喹啉甲瓦龙酸酯。Francotte等的研究还发现,SMB对于难溶的化合物,如formoterol尤为有用。而且可调节不同参数如进样率和萃取率来达到最佳纯度和产率。
领苯二甲酸酯(PAEs),是环境激素类的一种,作为聚氯乙烯、纤维素树脂、天然橡胶和合成橡胶的增塑剂广泛应用于塑料工业中,具有致畸、致突变、致癌以及生殖毒性。领苯二甲酸酯与塑料分子之间并不是以化学键结合,而是由氢键或范德华力连接,彼此保持各自独立的化学性质,因此它极易从塑料中释放,从而转入空气、水体、底质和生物等环境载体中,通过食物链危害着人体健康。领苯二甲酸酯类对水体的污染在国内已有许多报道,其主要来源于生产和使用邻苯二甲酸类的工厂所排放的工业污水。贝类属于滤食性生物,移动性相对较差,生活低于极为固定,所处环境受污染后,难免在滤食饵料时从水中吸入各种有害化学物质,PAEs等有机物具有较强的亲脂性,使其能在贝内中进行生物累积,并最终经食物链传递危害人类健康。因此建立一种高效、简便、准确的方法来检测贝类肉中领苯二甲酸酯类,对于开展贝类生物体中PAEs安全评估具有重要的指导意义。目前,国内外PAEs的分析方法主要有气相色谱法、液相色谱法及色谱-质谱联用法,分析的样品以环境、食品、塑料样品为主,尚未见对淡水贝类的研究报道。本文选取液相色谱法测定淡水贝类中8种领苯二甲酸酯的方法,该方法简便、快速,重现性好,准确度高,为淡水贝类中领苯二甲酸酯类物质准确的分析提供了理论参考。实验方法:1.样品的处理将购置好的新鲜贝类置于烧杯中,70度水浴加热30min,将贝类除壳以外所有贝肉全部取出,加入适量的生理盐水后,使用高速组织捣碎机,以10000r/min处理5min,然后移入50ml的离心管中,于4°C,15000r/min条件下离心15min,弃去上清液,将下层固体小烧杯中待用。称取贝肉5g,置于50ml离心管中,加入25ml1:1的正己烷和二氯甲烷混合液中,500W,53HZ超声提取30min后,在4°C下以15000r/min转速离心10min,移取上清液至旋转蒸发仪上浓缩,浓缩液移入2ml试管中,用甲醇定容到2ml,保存备用。测定时,提取液需经聚苯乙烯柱净化和0.45ul有机滤膜过滤后供HPLC测定,每个样品平行测定三次。2.标准溶液配制以甲醇为溶剂,配制浓度分别为0.1、0.5、1.5、10、25、50ul/ml的PAEs混合标准工作液。绘制标准曲线,用外标法计算样品中PAEs的含量。3.色谱条件色谱柱:AgelavenusilXBP-C18(250*4.6*5);流动相为甲醇-水梯度洗脱,检测器为紫外检测器,检测波长230nm,流速1.0ml/min,柱温25°C,进样体机1ul。结论本文以正己烷和二氯甲烷为提取剂,液液萃取贝类中的8种PAEs,以甲醇-水为流动相采用高效液相色谱仪建立了淡水贝类中领苯二甲酸酯类化合物的检测方法。方法操作简便,回收率高,重现性好。在实际样品分析中,;螺蛳,黄蚬和田螺中8种PAEs均有检出。
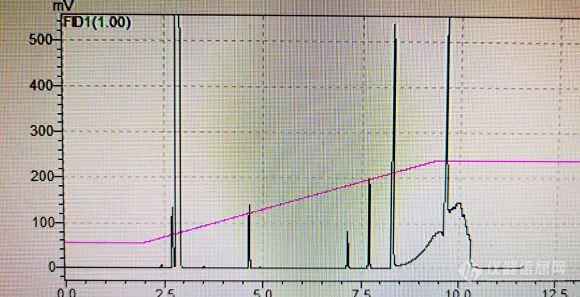
如题:最近有个胺类盐酸盐需要GC监控,试了一下饱和碳酸钠水溶液条件然后拿DCM萃出来检测,用的柱子原来是RTX-5,结果发现不止该化合物拖尾严重,其余杂质,原料等都拖尾严重,于是更换了色谱柱,手上除了RTX-5以外就只有WAX柱,RTX-1701的柱子,同规格的,后来选了RTX-1701,因为WAX柱子担心对其损伤太大,因此选择了1701,用该柱子定位了响应的化合物以及目标盐酸盐,所有化合物出峰都非常好,峰形对称,唯独目标化合物盐酸盐出峰很差,而且比RTX-5还要差。化合物结构如图:[img=,202,81]https://ng1.17img.cn/bbsfiles/images/2020/08/202008051729152242_37_3116636_3.jpg!w202x81.jpg[/img]化合物[url=https://insevent.instrument.com.cn/t/Mp]气相[/url]检测图如图:两种峰形都是该化合物,不同时间段的反应样品。[img=,580,297]https://ng1.17img.cn/bbsfiles/images/2020/08/202008051725102303_8716_3116636_3.jpg!w580x297.jpg[/img][img=,504,261]https://ng1.17img.cn/bbsfiles/images/2020/08/202008051725136944_4475_3116636_3.jpg!w504x261.jpg[/img]不知道问题出在哪里,这个情况是色谱柱不适用?只能使用胺类柱吗?我搜了一下类似的帖子,发现有个楼主发了一个胺类盐酸盐的分析方法思路,用的是2%NaOH甲醇溶液破坏盐酸盐,且用氮气保护,原因是裸露的氨基容易被氧化然后用DB-1的色谱柱进样,峰形良好。我在思考是否可以用同方法配样,但是氢氧化钠在[url=https://insevent.instrument.com.cn/t/Mp]气相[/url]中也是不能汽化的,这是否对色谱柱造成了影响,且碱性较大是否也会损坏柱子??
[color=#444444]安捷伦7890A[url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]测定甲乙丙丁戊5种醇类,顶空进样,溶剂是乙酸乙酯,使用的柱子是DB-624(30x0.25x0.14),进样口温度180,流量1.2,顶空瓶放入配置好的标准溶液10ul,甲醇峰经常出现平顶峰或者圆顶峰的现象,按照故障排除的方法,不断减小进样量和加大分流比,调试到峰比较正常一点的时候,信号值只有20PA左右,峰面积大概也只有100左右,这样正常吗?正常的时候,其它四个醇类的峰都比较正常,但是甲醇峰始终还是有一点拖尾,这是怎么回事呢?[/color]
我是做提取的,提取出来的东西中含有酯类、醇类和少量的烃类。现在想找分析方法,我准备用液相色谱做,用蒸发光散射检测器检测。但是在色谱柱和流动相的选择上比较疑惑。。。 应该是用正相色谱柱吧,但是用哪个型号呢?还有,我的这个东西常温下不太易于溶解于正己烷,只是在氯仿和四氯化碳中能稍微溶解一点,所以,我该选择什么作为流动相呢?希望大虾指教。。。。
前处理需要衍生。摘要:建立了[url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url],火焰光度检测器测定柑橘类水果中苯丁锡残留量的检测方法。样品在酸性条件下经丙酮及正己烷萃取并浓缩,用正己烷溶解残渣,经乙基溴化镁衍生后,采用硅胶固相萃取柱净化,正己烷,二氯甲烷( 体积比为*. $)混合液洗脱,用毛细管[url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url],火焰光度检测器( 锡滤光片:610nm)测定,外标法定量。结果表明:该方法的线性范围为0.2-2.0mg/l,相关系数r=0.9995;当阴性脐橙样品中加标水平为0.1-0.4mg/kg时,苯丁锡的回收率为79.6%-109.6%,相对标准偏差为3.6%-9.05%,方法的检出限为0.1mg/kg。该方法重复性好,灵敏度高,完全满足国内外柑橘类水果中苯丁锡残留分析的要求。
有木有做HPLC核苷类成分分析的,都用的哪家的哪个型号的色谱柱呀?求告知一下,谢谢
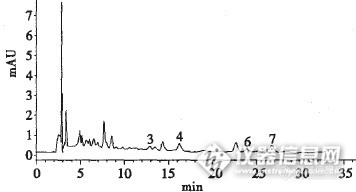
液相色谱测定土壤中的苯胺类污染物摘要:试验建立了土壤中苯胺类污染物的液相色谱测定方法,方法线性关系很好,7种苯胺类成分的检出限在0.1ug/kg-0.5 ug/kg之间,空白加标试验的相对标准偏差RSD在1.9% -4.9%之间,基质加标回收率在64.1% ~92.8% 之间。实际样品的测定结果表明该方法分离效果好,能够满足土壤中苯胺类分析的要求。土壤污染是当前人类面临的一个极为重要的、全球性的环境问题之一。土壤中污染物主要包括重金属、有机污染物、放射性物质、过量的氮(N)、磷(P)以及病原菌等,它们对粮食安全和人类生存环境产生胁迫。其中,苯胺类化合物是土壤中常有的有机污染物。苯胺类化合物常见于印染、制药等土壤中,苯胺类是一种重要的有机化工原料,环境中的苯胺主要来源于橡胶、印染、制药、塑料、陶器上釉等工艺生产过程。环境中苯胺的残余量已引起人们的极大关注,早已将其列人优先监测的污染物。其中,美国、日本等国把苯胺类列入主要监测项目或优先监测污染物的黑名单 。因此,建立简单、快速、灵敏、有效的苯胺类测定方法十分必要。苯胺、硝基苯胺和二硝基苯胺有3种异构体:邻硝基苯胺、间硝基苯胺、对硝基苯胺、2,4一二硝基苯胺、2,6一二硝基苯胺、3,5一二硝基苯胺等是印染、橡胶、制药、塑料和油漆等的原料,是染料工业的中间体。这几种化合物可通过呼吸道、消化道而摄人体内,使氧和血红蛋白变为高铁血红蛋白影响组织细胞供氧而造成内窒息,且被认为对人体有很强的致癌性。因此,苯胺类化合物对环境的污染一直被人们所关注,在我国苯胺类化合物也被列为环境中的重点污染物,并制定了最高容许排放浓度。苯胺类化合物的测定在国家环保局所编的《水和土壤监测分析方法》一书中是采用“萘乙二胺偶氮光度法”,该方法具有简便、精密度和准确度好的优点,不足的是该方法只能分析总的苯胺类化合物,无法确定每种化合物的含量,而液相色谱就弥补了此不足,可以快速、准确的测定每种化合物的含量。在《水和土壤监测分析方法》第四版中采用的苯胺类液相色谱法C中,使用的是乙酸铵缓冲液和甲醇做流动相,能测定5种苯胺类污染物。1998年李瑞琴用69%正己烷、30%二氯甲烷和1%异丙醇做流动相测定了苯胺、邻硝基苯胺、间硝基苯胺、对硝基苯胺等4种苯胺类污染物。1 实验部分1.1 仪器和试剂1.1.1 仪器美国Agilent公司的1200液相色谱仪; 色谱柱:Ultimate XB-C18 4.60×250mm,5 um .Part Number 00201-31043 Serial Number 211302350.1.1.2 试剂1) 苯胺、邻硝基苯胺、间硝基苯胺、对硝基苯胺、2,4一二硝基苯胺、2,6一二硝基苯胺、3,5一二硝基苯胺为色谱纯标样。2) 二氯甲烷、丙酮、乙睛等均为色谱纯试剂。3) 氢氧化钠、盐酸均为分析纯试剂。4) 无水硫酸钠为分析纯试剂,用前在马弗炉中350烘4 h。5) 氟罗里硅土100~200目色层分析用,进口分装,400℃下烘2h,置干燥器中备用。1.2 样品制备1.2.1 土样预处理将土壤样品风干、研磨后过筛,称取土壤20g,加适量硅藻土拌匀,填人萃取池中,若萃取池中仍有空间,则用硅藻土填满,萃取。取下样品萃取液,于38℃用氮吹仪进行浓缩至lmL,待净化。1.2.2 样品净化苯胺类的预分离和浓缩:称取含水量10%的氟罗里硅土6g,制成环巳烷浆液,湿法装入内径为lOmm的玻璃柱中,新装好的色谱柱应用二氯甲烷和丙酮冲洗数次,然后将预浓缩过的萃取液通过此玻璃柱,再用2mL的环巳烷洗K—D浓缩瓶三次并通过层析柱,用30mL二氯甲丙酮洗脱液浸泡层析柱,再用30mL洗脱液洗脱柱中苯胺类,全部洗脱液接入氮吹瓶中于氮吹仪中浓缩定
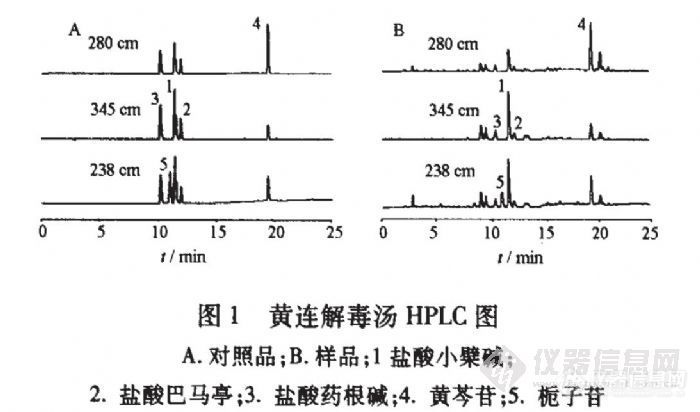
多波长高效液相色谱法同时测定黄连解毒汤中3类成分李新中1,雷鹏,刘韶(中南大学湘雅医院,湖南长沙410008) 目的:同时测定黄连解毒汤中3类有效成分(盐酸小檗碱、盐酸巴马亭、盐酸药根碱、黄芩苷、栀子苷)的含量。方法:三波长同时检测的高效液相色谱法;Diamonsil C18。色谱柱(4.6 mm×250 mm,5um);流动相水-甲醇旬.05%磷酸(梯度洗脱);柱温35℃;流速1 mL·min~;检测波长345,280,238 am。结果:建立了同时对黄连解毒汤中的5个成分进行定量的测定方法,本法快速、重复性好、灵敏度高。结论:为黄连解毒汤提供了更合理、可靠的质控方法。http://ng1.17img.cn/bbsfiles/images/2012/07/201207232347_379320_2355529_3.jpg
最近在开展氨基糖苷类(包括卡那霉素\庆大霉素\链霉素\双氢链霉素\新霉素\安普霉素等)的检查,虽然有国标使用c18在做,但考虑到七氟丁酸容易污染到质谱,很是怕怕,所以就选择了hilic色谱柱。第一次接触hilic色谱柱,遇到了以下问题:现象:1. 使用等度洗脱,乙腈:水=90:10,所有项目的保留时间都在2.6min附近。2. 使用梯度洗脱,流动相还是乙腈:水,每个项目出现了两个保留时间,一个还是在2.6min附近,另一个在9min附近,两个保留时间都是在乙腈比例最高的时候出现。3.怀疑出现的峰是色谱柱被洗脱掉的物质,使用空白水进行测试(以上标准品使用水溶解),未出峰。排除是色谱柱及液相系统的问题,说明这些峰应该是想要的标准品峰。4.从论坛资料查询到的,有人说是用乙酸铵或甲酸铵可以,经测试,加入铵后,标准品不出峰或惨不忍睹。我使用的是10mM的浓度,不清楚是否是浓度太高的问题,但我觉得可能性不大。疑问:1。所有的标准品保留时间一样,非常奇怪,很是怀疑这到底是不是我要的标准品峰。2。跑标准曲线,不成线性,甚至部分项目低浓度的感度高于高浓度的,郁闷。。。3。保留时间无法再向后拖了,因为乙腈的比例够高了,应该再怎样改变保留时间呢?自己也搜索了一些资料,可是还没有一个比较完善的关于HILIC色谱柱的使用方法的资料,论坛中有人说使用HILIC是可以分析这些物质的,但给出的资料或说明又难以让人信服。所以,希望对HILIC了解或正在使用HILIC的大侠多多给点意见,如果有人确实在做这几种物质的话(使用HILIC),小生那更是感激不尽了。。。。欢迎大家在此热烈讨论。。。更新:在论坛上搜到了个HILIC方法开展指南,给大家分享下。欢迎继续探讨,希望这个帖子能够成为hilic开展方法的探讨贴。。。更新: 看了些资料,hilic色谱柱使用需要以下注意事项:1。流动相至少需要5%的极性溶剂(如水,甲醇等)2。流动相需要不低于40%的有机相(如乙腈)3。使用甲酸铵或乙酸铵能得到较好的重现性。4。定溶液不能使用100%的水5。平衡时间比较长,一般需要10倍以上的柱体积平衡更新: 经过一段时间的测试,通过使用hilic色谱柱目前还没有得到良好的效果。使用资生堂的一款链霉素专用色谱柱,保留特性不错,只是感度还有点差,需要再进行优化。由于最近忙于粘杆菌素的开展,氨基糖苷类的测试需要拖一拖了,如果,谁也在做粘杆菌素,不妨也来讨论下。
[color=#444444]之前查看文献检测卵磷脂类物质用C18柱检测,照着文献检测后发现文献纯粹是瞎扯。后来经查阅英文文献和博士论文,确定了一个比较靠谱的方法。使用的是正相硅胶柱,流动相以氯仿为主(我也不想用氯仿,但文献上使用该方法检测出来的峰确实很好,各物质都分开了),实验室目前买不到色谱级的氯仿(就目前用的几瓶分析纯的氯仿还是费老劲买的,又是备案又是调查的),本来想把氯仿旋蒸的,网上搜索后发现旋蒸氯仿一是回收率不高(氯仿挥发太快不易回收)二是毒性太大(旋蒸时挥发的满屋子都是)。我的问题是:各位有遇到我类似问题的嘛?怎么解决?我直接用分析纯的氯仿上柱子吗?[/color]







 我要推广仪器
我要推广仪器
 下载APP
下载APP