[color=#444444]两个极性相近的物质,想要调整实验条件,把色谱峰分离开来,流动相乙腈,0.1%的甲酸水溶液,C18柱,[color=#444444]分离不好,可以分的开,就是两个峰很近,保留时间相差0.8min。[/color][/color]
由【求助】测有关物质色谱条件的专属性试验问题 http://bbs.instrument.com.cn/shtml/20101025/2882558/想到的1、两杂质色谱峰达到基线分离时的峰面积之和与两色谱峰部分重合时的峰面积之和,在使用自身对照法时它们所占的比例是一样的吗?2、用于校正归一法时,我觉得两峰应该完全分离,你觉得的呢?
[color=#444444]液相色谱改变流动相配比后需要重新走基线吗?液相峰分不开可以采取哪些措施来分离?[/color]
怎样使黄酮色谱峰分离更好?
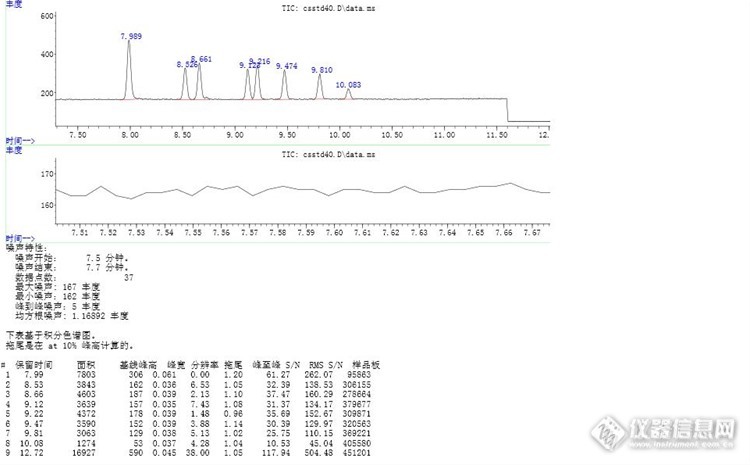
使用安捷伦GCMS新的软件计算色谱峰的分离度,但是不知道如何看懂报告,请老师指点!请老师指出各个色谱峰的分离度,其中Rthttp://ng1.17img.cn/bbsfiles/images/2017/03/201703221726_01_1794257_3.jpg12.7min的峰没有截图出来。
请大家说说一般两个主色谱峰分离不开来可以采取哪些方法?
各位专家、学者,大家好,最近想分离N2,H2,CO,CH4,CO2,C2H4,C2H6几种气体,用的是GDX-502色谱柱和5A分子筛色谱柱,氩气作载气,请问一下大家,柱温,检测器温度,柱流速大约多少,分离效果好点,由于没有经验,一直在调试,效果不太好,还有就是柱温以及载气流速分别影响气体分离的那些方面。比如出峰时间、峰型等。谢谢各位专家热心的帮助。
我现在在做用RP-HPLC分离蛋白质,分离效果一直不理想(见谱图),谱图中的流动相比例是目前最好的,但是目标峰与前面的杂峰分离不开,达不到基线分离,希望各位高手可以指导一下,十分谢谢!色谱条件:ultimate XB-C18色谱柱(4.6×250mm,300A);流动相是0.1%三氟乙酸水和0.1%三氟乙酸乙腈;检测波长228nm,采用梯度洗脱。http://ng1.17img.cn/bbsfiles/images/2011/08/201108201550_311298_2246796_3.gif
各位,请教下如何计算色谱峰的分离度,我用的是戴安的ICS-3000/5000,软件上能实现吗?还有,分离度R大于多少时说明两峰完全分离?
三月份的时候用TG—5MS 弱极性柱子打的乙醇,峰的分离看上去还行,可前几天再去打了一遍,发现峰分离不开,色谱条件都没改,唯独是换了衬管,之前的衬管是分流/不分流衬管,,后面换的是分流直式衬管,结果发现甲醇和乙醇峰分不开,后面再把之前用的那个衬管换上,可以分离出来,可分离得不好,改了进样口温度为170度,其余条件不变后,稍微好点,附上图谱,和原色谱条件 进样口温度200 , 分流流量 50 ,柱流量1 ,分流比50:1,吹扫3;检测器220℃,空气400, 氢气 40 ,尾吹30。 图一为三月打的色谱,图二为三月份用的衬管,图三为最近打的色谱,图四为最近用的衬管,图五为最近用三月份的衬管和修改了进样口为170摄氏度打的色谱。如果弱极性柱子能分离出乙醇甲醇峰,还有没有必要用强极性柱子,这些色谱图都是用弱极性柱子打的,以及换了衬管后分离不出峰,是不是分流比不对还是衬管不合适的问题,用了直通式的衬管打的,峰都是分离不开。[img]https://ng1.17img.cn/bbsfiles/images/2020/06/202006101744320408_8756_4115190_3.png[/img][img]https://ng1.17img.cn/bbsfiles/images/2020/06/202006101744321453_8101_4115190_3.png[/img][img]https://ng1.17img.cn/bbsfiles/images/2020/06/202006101744325997_7107_4115190_3.png[/img][img]https://ng1.17img.cn/bbsfiles/images/2020/06/202006101744330020_1747_4115190_3.png[/img][img]https://ng1.17img.cn/bbsfiles/images/2020/06/202006101744341085_5576_4115190_3.png[/img]
[color=#444444][url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]两个峰看上去已经部分重合了,但是分离度却大于1.5,这是算完全分开吗?还有就是数据处理的时候需要扣除空白(溶剂)吗?[/color]
液相色谱分离的峰,每一个峰都只是代表一种化合物吗?还是有可能是几种化合物的混合?峰没有分开是因为流速大还是流速小?流速是怎么影响峰的?谢谢。
如何判断色谱峰分离程度的好坏,是否有一个标准呢?紧急求助,谢谢各位!
液相色谱大峰前小峰怎么分离[img]https://ng1.17img.cn/bbsfiles/images/2019/08/201908061642384749_7239_3963683_3.png[/img]
最近刚买了一台[url=https://insevent.instrument.com.cn/t/Mp]气相[/url],采用的程序升温方法和我们正在使用的GCMS一样,进同样的样品,[url=https://insevent.instrument.com.cn/t/Mp]气相[/url]的峰却无法分离,尝试了一下改变程序升温,还是无法分离,请问是什么原因呢?还想请教一下大家程序升温的方法设定大家都是怎么判断和设置的呢?[img]https://simg.instrument.com.cn/bbs/images/default/em09512.gif[/img]我们现在使用的是安捷伦的非极性色谱柱
对于定性分析,色谱峰的分离度应至少达到多少呢?
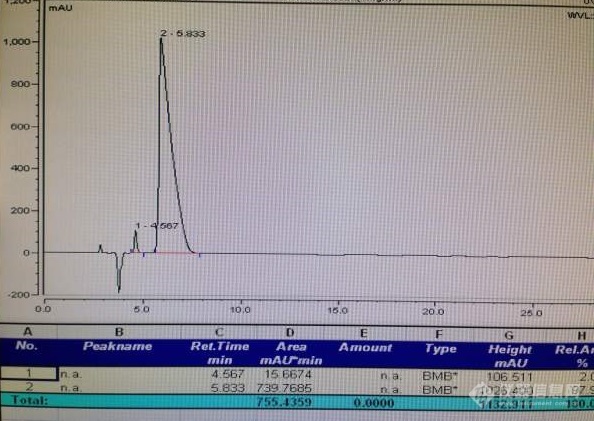
[color=#444444]现想求助一色谱分离问题。[/color][color=#444444] 我做液相色谱时,有两个峰保留时间较近,想让他们分开,大家有什么好方法吗,我已经调过流速、流动相比例和柱温了效果都不明显,具体色谱条件如下,色谱柱:Agilent Eclipse Plus C18(3.5um,4.6*100mm);流动相:0.1%甲酸水:甲醇(95:5),等度;柱温:30℃;流速0.3mL/min;液相色谱图见附件,保留时间为4.567的为图片中的手写结构式,保留时间为5.833的为另一结构式,我想让这两个峰的分离时间达到两分钟以上,因为想让前一个峰进质谱,后一个峰不进,如果浓度降低一起进质谱的话前一个峰就没什么响应了,求高手赐教有什么好办法让这两个峰分的更开一点,谢谢![/color][color=#444444][img=,600,450]https://ng1.17img.cn/bbsfiles/images/2019/09/201909231012429519_6927_1646718_3.jpg!w600x450.jpg[/img][img=,594,421]https://ng1.17img.cn/bbsfiles/images/2019/09/201909231012406386_9342_1646718_3.jpg!w594x421.jpg[/img][/color]
GDX101色谱柱测乙炔,氯乙烯气体。最近几天此2峰不能完全分离,且拖尾严重,条件未变。以前都可以分离开,为啥现在不行了呢?请各位大虾指点,是柱子有问题了还是怎的?
当色谱峰出现重叠,改变硬件条件的情况下如果还不能解决。 可以用卡尔曼滤波算法进行软件分离而得到准确的面积。http://ng1.17img.cn/bbsfiles/images/2017/03/201703071038_01_1780790_3.png上图是一个分离效果图。通过众多的样品测试分析,卡尔曼滤波算法对于重叠程度低于90%的,分离效果相当好,对于重叠程度大于90%而小于95%的有一定误差,而超过95%的基本就无能为力了。色谱分析中经常出现基线漂移,出峰时间是变动的。这也可以通过采用一定软件算法进行校正。在校正前需要指定一个正常的谱,然后通过神经网络算法进行学习。测试其它样品时如果发现有漂移就会计算出漂移距离(时间),再进行校正。以上重叠峰分离方法和漂移校正方法同样适合光谱分析。如果需要技术交流的,可点击头像联系
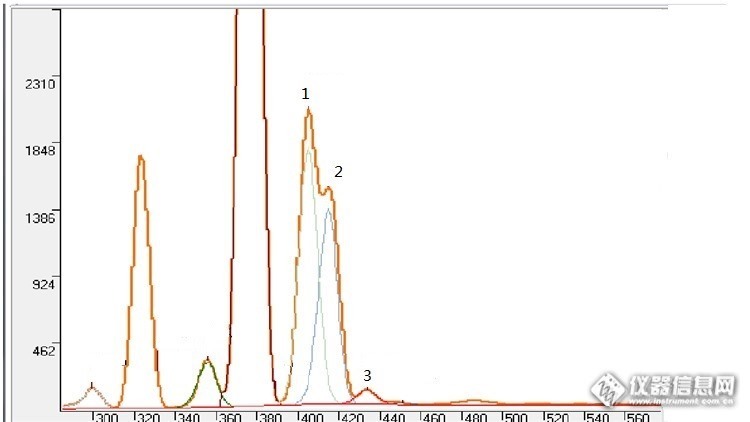
当色谱峰出现重叠,改变硬件条件的情况下如果还不能解决。可以用卡尔曼滤波算法进行软件分离而得到准确的面积。http://ng1.17img.cn/bbsfiles/images/2017/03/201703071038_01_1780790_3.png上图是一个分离效果图。通过众多的样品测试分析,卡尔曼滤波算法对于重叠程度低于90%的,分离效果相当好,对于重叠程度大于90%而小于95%的有一定误差,而超过95%的基本就无能为力了。色谱分析中经常出现基线漂移,出峰时间是变动的。这也可以通过采用一定软件算法进行校正。在校正前需要指定一个正常的谱,然后通过神经网络算法进行学习。测试其它样品时如果发现有漂移就会计算出漂移距离(时间),再进行校正。以上重叠峰分离方法和漂移校正方法同样适合光谱分析。如果需要技术交流的,可联系我 电话:13926563756 qq:648048428 Email: midstone@126.com
不锈钢色谱柱管为什么会引起色谱柱分离组分峰形拖尾?请给指点
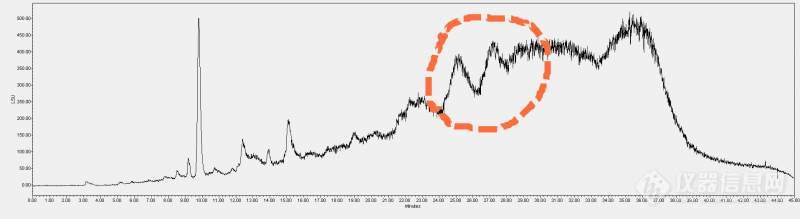
[color=#444444]在做糖的分离,用的是HILIC柱,流动相是乙腈和甲酸-甲酸铵缓冲液[/color][color=#444444]流动相比例为 0 : 80% (乙腈)+20%(缓冲液)[/color][color=#444444] 30: 60%(乙腈)+40%(缓冲液)[/color][color=#444444] 40: 10% (乙腈)+90%(缓冲液)[/color][color=#444444]色谱图如下:[/color][color=#444444]怎样调节流动相才能让虚线部分的峰更好一点呢?求大神指教,万分感激[/color][color=#444444][img=,690,188]https://ng1.17img.cn/bbsfiles/images/2019/08/201908121348556006_4126_1701336_3.jpg!w690x188.jpg[/img][/color]
两种标准品,色谱峰有重叠,请问有什么方法可以分离吗?我不想改变流动相的配比,改变流速是不是可以呢?[em0906]柱温35,254nm,流动相甲醇:乙酸=45:0.2%
‘有奖问答’选择题:在色谱的流出曲线上,相邻两组分色谱峰的分离度主要取决于( )A. 分配系数 B. 保留参数 C. 色谱柱温度 D. 载气流速
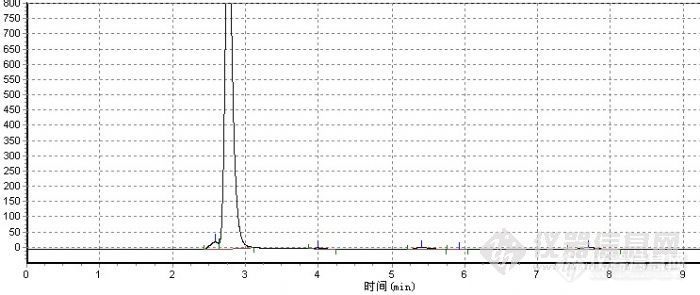
液相色谱检测条件:波长:365;柱温:40℃;流动相:甲醇;流速:0.8;灵敏度0.01; 称重:0.015g;溶剂:100ml三氯甲烷; 进样量:5ul.http://ng1.17img.cn/bbsfiles/images/2010/10/201010151639_251672_2166859_3.jpg各位我现在想请教你们一个问题,我这个主峰是在2.8几出峰的,我现在想他往后推迟,与溶剂峰分开应该怎么做?而且该样品应该有2个峰,该怎么分离出,条件怎么改··谢谢~~!
工作站中,都会自动给出分离度的数据。分离度的定义是“相邻两峰保留时间差与半峰宽的比值”,那么:1》假如一个色谱图,有连续三个峰,第二个峰的分离度,是以peak 1 和peak 2 还是peak 2与peak 3的数据计算?2》不管是与前一个还是后一个峰的数据计算,都会导致两端一个峰没有分离度数据,这样我觉得是不合理的。3》依据经验,peak 2 是以peak 1 和peak 2的数据来算的,假如peak 2和peak 1分离得开,但是和peak 3分离得不是理想,那么,peak 2的分离度即使大于1.5,并不代表其与相邻峰分开........欢迎大家讨论
各位有色谱方法开发经验的前辈:现有两个问题需请教1.对于高浓度主峰,峰宽很宽,有6分钟样子,在紧随其后有1杂质峰,与主峰分离度只有0.8,色谱条件除了色谱柱可以更换外,其他条件均不能改变,那么用何种色谱柱可以提高两者的分离度为1.5以上;2.哪类色谱柱更适合于分析高浓度化合物;谢谢各位赐教,不胜感激!
请问以石油醚为溶剂的联苯菊酯应该以什么色谱柱分离才会出峰? 我们这里是安捷伦气象色谱仪,柱子是ov1701,FID检测器。。 试了好几次都只出一个石油醚的峰。。。很着急,一直检测不出联苯菊酯。。 请高手帮忙分析一下。在这种条件下,各项参数应该设为多少才能分理处联苯菊酯???是柱子的问题还是检测器的问题?? PS:我的样品室2mg/L 最大浓度也不超过100mg/L.是不是因为检测器不够灵敏导致无法出峰?但是我查了有些文献联苯菊酯也是用FID检测器测的,浓度跟我的差不多。 请高手帮帮忙。调了好多次参数都不能出结果。。很着急。。在线等。。。。 问题补充: 因为我在做微生物农药降解,最大样品浓度也不超过200mg/L,昨天试了一个1g/L的标准品还是不能出峰,程序升温到280度,时间拖到6分钟只看到一个石油醚的峰。分流比采用10:1.昨天又测了一下石油醚标准品,发现跟加过联苯菊酯后的峰一模一样,感觉不像是因为溶剂跟溶质混溶导致只出一个峰,应该是检测器不够灵敏检测不出来低浓度的样品吧。 我想改成高效液相色谱试试。。但是液相我从来没用过,还在研究中。
色谱图上没有完全分离的峰如何积分,积分规则是什么
在液相色谱中产品峰哈杂质峰分不开,怎么办?有哪些因素影响分离度?







 我要推广仪器
我要推广仪器
 下载APP
下载APP