用半定量薄层色谱法,按照GB/T 8381-2008测饲料中黄曲霉毒素B1 在标准中提到展开剂:使用带盖展开槽,展开槽内壁衬上吸水纸,使展开槽被展开剂饱和;三氯甲烷-丙酮混合液(90+10):在未饱和展开槽内,90ml三氯甲烷和10ml丙酮混合;乙醚-甲醇-水(94+4.5+1.5)混合液:在饱和展开槽内,94ml乙醚、4.5ml甲醇和1.5ml水。那么在这里如何确定展开槽是饱和的还是未饱和的,怎么进行操作呢?
大家谁有用过水平展开槽的,10*10左右。哪里能买到。之前有用过,不过现在找不到那个供应商了。很简易的一个,上面只需盖上一个玻璃片就好。空间小,饱和快。
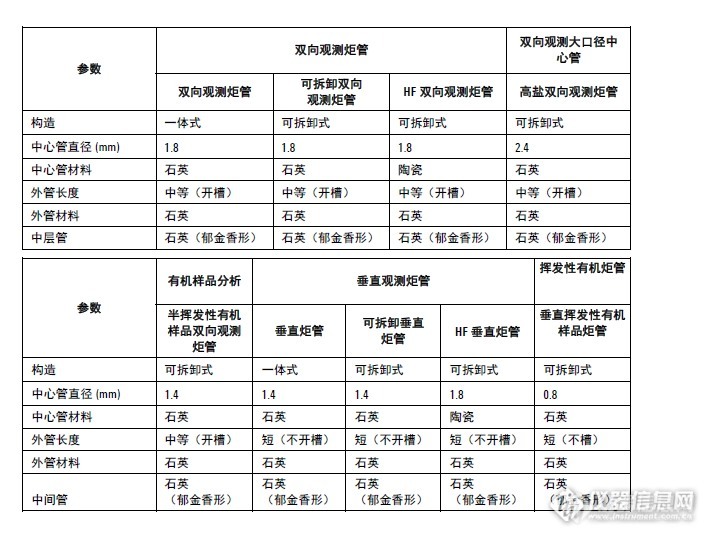
[img=,690,522]http://ng1.17img.cn/bbsfiles/images/2017/05/201705082301_01_2140715_3.png[/img]如图片介绍,炬管的外侧开槽和不开槽有哪些区别?
公司以姜黄为主,最近做姜黄的薄层色谱,一切按药典上来,配制展开剂,三氯甲烷:甲醇:甲酸=96:4:0.7我配好后震摇混匀,倒入薄层色谱展开槽,密闭展开槽饱和半小时后放入样品板,样品跑的速度不一样,有的快有的慢,都没在一条线上,以前也常有这个问题,做好几个只有一个比较完美的。展开剂是现配的,震摇过,但是我发现放置后有点浑浊,再次震摇才能澄清,不知是否与温度有关系,目前放展开槽的地方没有开空调,温度约10-15℃左右。样品板是20*20的,以前用的是10*20 的。求解答,如何解决这类问题?是否与展开剂有关?以下图片为还在展开槽中的样品,高低明显差异……[img]https://ng1.17img.cn/bbsfiles/images/2019/01/201901151403474512_9091_3116636_3.png[/img]
怎样确定冲击试样的缺口开槽方向?即在原材料的那个位置,加工时候怎样标记呢?
最近我在做TLC,但是不管怎么调试展开剂,还是出现拖尾,不知道为什么,哪位高手帮帮忙 PS:我是在展开剂和控湿剂同时饱和15min之后,立即将点好样的merck板放进展开槽展开,不知道这样做对不对~~
请教各位高手,为什么在一个展开槽里面展开的两张板的结果总是相差很远,究竟是什么原因,最近好让人郁闷的~~
薄层色谱法(简称TLC)。真如“省部重点实验室”所言:薄层色谱实际上是柱色谱的一种改良,薄层板可以认为是一个开放的色谱柱。但就技术操作来看,又很类似纸色谱。其操作方法概述如下:先制备薄层板,即在大小适当的玻璃板上,均匀涂上吸附剂,厚度在一毫米以内,然后在距底边1.5厘米处点上样品溶液,形成一个小点,称为“原点”。再将薄层板置于盛有动相溶剂的玻缸内(此溶剂称为“展开溶剂”,玻缸称为“展开槽”)。当溶剂沿薄层扩散到距原点以上一定距离时(一般10—12厘米),取出薄层板,记录展开溶剂扩展前沿距原点的距离A。然后用喷洒显色试剂或紫外光线照射的方法使被分离的化合物显色,此过程称为“显谱”。但由于不同的样品需要使用不同的显色方法,故对于标准仪器的要求显得就凌乱了。TQ-1是最早也是较为全面的薄层色谱法启动包。但是怎么样才能真正组合一套薄层色谱仪,用户拿来就能用,既能加热显色,又能喷雾显色,也能紫外检测。考虑成本关系,扫描仪就别考虑了。这可是个世界性难题。欢迎专家学者来组合一个行业标准的启动包---薄层色谱仪
薄层色谱又叫薄板层析,是色谱法中的一种,是快速分离和定性分析少量物质的一种很重要的实验技术,属固—液吸附色谱,它兼备了柱色谱和纸色谱的优点,一方面适用于少量样品(几到几微克,甚至0.01微克)的分离;另一方面在制作薄层板时,把吸附层加厚加大,因此,又可用来精制样品,此法特别适用于挥发性较小或较高温度易发生变化而不能用[url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]分析的质。此外,薄层色谱法还可用来跟踪有机反应及进行柱色谱之前的一种“预试”。薄层色谱(Thin Layer Chromatography)常用TLC表示,又称薄层层析,属于固-液吸附色谱。是近年来发展起来的一种微量、快速而简单的色谱法,它兼备了柱色谱和纸色谱的优点。一方面适用于小量样品(几到几十微克,甚至0.01μg)的分离;另一方面若在制作薄层板时,把吸附层加厚,将样品点成一条线,则可分离多达500mg的样品。因此又可用来精制样品。故此法特别适用于挥发性较小或在较高温度易发生变化而不能用[url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]分析的物资。此外,在进行化学反应时,常利用薄层色谱观察原料斑点的逐步消失来判断反应是否完成。 薄层色谱是在被洗涤干净的玻板(10×3cm左右)上均匀的涂一层吸附剂或支持剂,带干燥、活化后将样品溶液用管口平整的毛细管滴加于离薄层板一端约1cm处的起点线上,凉干或吹干后置薄层板于盛有展开剂的展开槽内,浸入深度为0.5cm。待展开剂前沿离顶端约1cm附近时,将色谱板取出,干燥后喷以显色剂,或在紫外灯下显色。记下原点至主斑点中心及展开剂前沿的距离,计算比移值(Rf):

按方法,展开剂氯仿-乙酸乙酯-甲醇-水=15-40-22-10的配比在冰箱里冰完取下层,展开然后是用双杠,一边是57硫酸+100水的硫酸溶液,一边是展开剂。有预饱和板在点完之后,有去真空干燥箱里用五氧化二磷常温干燥过夜。展开的时候,屡次出现展开剂分层(室温28度),下面展开剂乱七八糟的展开前沿(忘记拍照了)后面听从网友帮忙,说温度低点会好很多,我撤去了硫酸溶液,,展开槽空调对着吹(17度),然后爬出来还是分层,但好多了[img=,230,408]http://ng1.17img.cn/bbsfiles/images/2017/05/201705041356_01_2240076_3.jpg[/img]然后后面还是颜色了,但是展开的很差用的是青岛海洋的硅胶G[img=,230,306]http://ng1.17img.cn/bbsfiles/images/2017/05/201705041358_01_2240076_3.jpg[/img]一个一个点胖墩墩的。没分离好。求网友帮忙指导下。
单向二次展开薄层色谱法单向二次展开薄层色谱法检识挥发油中各成分时,为什么第一次展开所用的展开剂极性最好大于第二次展开所用的展开剂的极性?单向二次展开薄层色谱法有什么优点?
中药材薄层色谱展开系统之浅见 工作几年以来,主要运用薄层色谱分离方法对中药材成分进行分析。薄层色谱直观的色彩图像表达方式是一大特点,通过各种展开剂的灵活运用可将中药材分成不同极性组分进行分离,充分展现出层析分离的意义。根据这几年的摸爬滚打,反复实践,总结出一点展开系统的经验。由于接触的药材有限,有些仍不够深入,因此观点可能有些片面,鄙薄之处望多多包涵!说明:以下使用的板为硅胶正相薄层板 1 黄酮类经典展开剂:甲苯-乙酸乙酯-甲酸(5:4:1),该系统还可用于香豆素类,三萜类成分分离,比例应作适当调整。 2 正己烷-乙酸乙酯或环己烷-乙酸乙酯系统,可分离极性相对较小的系统,如单萜类,木脂素类成分。可适当加甲酸,因己烷系统易使斑点扩散,甲酸对酸性成分分离有帮助。 3 乙酸乙酯-甲酸-冰醋酸-水(15:1:1:2)是甘草的展开系统,但其适用性很广,可用于中等极性的成分如绿原酸,DCQ,黄芩苷等。人参系统氯仿-乙酸乙酯-甲醇-水(15:40:22:10)(冰箱分层取上层溶液)也是一个不错的分离中等极性的展开剂。 4 若改变展开系统对分离帮助不大时,可考虑用变性板,如0.5%或1%NaOH溶液,0.1M NaH2PO4,0.1M Na2HPO4等。变性板还可调整斑点的Rf值,可将其减低。 5 氯仿是药典和文献广泛使用的展开溶剂,由于毒性较大,现在正逐渐用其他溶剂替代。有氯仿的两相系统易产生边缘效应,这与其沸点较低易挥发有关,若在其中加入些许酸或碱对分离可能产生意想不到的结果,特别是中药复方制剂时排除阴性干扰时,用量视情况而定。 总之,展开系统的千变万化使薄层色谱有了更多可操作可摸索的空间,因此有了更为丰富多彩的色谱图像,若能结合其它色谱得知化学成分的一一归属关系就更加深刻了。
1.原理试样酸化后,用乙醚提取苯甲酸、山梨酸。将试样提取液浓缩,点于聚酰胺薄层板上,展开。显色后,根据薄层板上苯甲酸、山梨酸的比移值与标准比较定性,并可进行概略定量。2.试剂异丙醇、正丁醇、石油醚(沸程30~60℃)、乙醚(不含过氧化物)、氨水、无水乙醇、聚酰胺粉(200目)、盐酸(1+1,取100mL盐酸,加水稀释至200mL、氯化钠酸性溶液。展开剂:正丁醇+氨水+无水乙醇(7+1+2)或异丙醇+氨水+无水乙醇(7+1+2)。山梨酸标准溶液:准确称取O.2000g山梨酸,用少量乙醇溶解后移入100mL容量瓶中,并稀释至刻度,此溶液lmL相当于2.0mg山梨酸。苯甲酸标准溶液:准确称取0.2000g苯甲酸,用少量乙醇溶解后移入100mL容量瓶中,并稀释至刻度,此溶液1mL相当于2.0mg苯甲酸。显色剂:溴甲酚紫-乙醇(50%)溶液(0.4g/L),用氢氧化钠溶液(4g/L)调至pH 8。3.仪器吹风机、色谱分离缸、玻璃板(10cm×18cm)、微量注射器(10μL,100μL)、喷雾器。4.分析步骤(1)试样提取称取2.50g事先混合均匀的试样,置于25mL具塞量简中,加0.5mL盐酸(1+1)酸化,用15mL、10mL乙醚提取两次,每次振摇1min,将上层醚提取液吸入另一个25mL具塞量筒中,合并乙醚提取液。用3mL氯化钠酸性溶液(40g/L)洗涤两次,静置15min,用滴管将乙醚层通过无水硫酸钠滤入25mL容量瓶中,加乙醚至刻度,混匀,吸取10.0mL乙醚提取液分两次置于10mL具塞离心管中,在约40℃的水浴上挥干,加入0.10mL乙醇溶解残渣,备用。(2)测定 聚酰胺粉板的制备:称取1.6g聚酰胺粉,加0.4g可溶性淀粉,加约15mL水,研磨3~5min,立即倒入涂布器内制成10cm×18cm、厚度0.3mm的薄层板两块,室温干燥后,于80℃干燥1h,取出,置于干燥器中保存。点样:在薄层板下端2cm的基线上,用微量注射器点1μL、2μL试样液,同时各点1μL、2μL山梨酸、苯甲酸标准溶液。展开与显色:将点样后的薄层板放入预先盛有展开剂的展开槽内,展开槽周围贴有滤纸,待溶剂前沿上展至10cm,取出挥干,喷显色剂,斑点成黄色,背景为蓝色。试样中所含山梨酸、苯甲酸的量与标准斑点比较定量(山梨酸、苯甲酸的比移值依次为0.82,0.73)。5.结果计算试样中苯甲酸或山梨酸的含量按下式进行计算。X=( A×1000)/m××1000式中,X为试样中山梨酸或苯甲酸的含量,g/kg;A为测定用试样液中山梨酸或苯甲酸的质量,mg;V1为加入乙醇的体积,mL;V2为测定时点样的体积,mL;m为试样的质量,g;10为测定时吸取乙醚提取液的体积,mL;25为试样乙醚提取液的总体积,mL。
为满足石化行业原油、渣油、蜡油、润滑油、重质油中饱和烃、芳烃、胶质、沥青质四组分分析要求,通过优化RY-CF19棒状薄层色谱仪配置及分析条件,完全可以满足此分析要求,其仪器具体要求如下所列:1. 能够检测原油、重质油、渣油、蜡油、润滑油四组分。2.恒温恒湿箱:温度范围5℃-300℃、控温精度0.1℃、容积不小于53L、带分段编程的控制器。3.扩展槽:能够将吸附有样品的色谱棒架置于展开槽中,在溶剂扩展的条件下,样品被有效地分离。4.操作方式:能够实现一次性机外送棒,自动扫描,计算机自动采集存储数据。5.色谱棒:色谱棒通过简单的再活化可重复使用50~100次,展开过程仅需少量溶剂。6.数据处理系统:专用软件包,控制薄层色谱分析仪工作,一个样品检测数据处理完毕后,自动启动下一个样品的检测,实时采集检测结果,并在薄层色谱专用软件中标绘出实时峰图,通过各峰图面积计算样品各组分的相对含量。7.燃气和助燃气:氢气、空气;流量控制系统:能够稳定控制氢气、空气流量。8.检测器:氢火焰离子检测器(FID),检测速度满足: 25、30 、35 、40、50、60 秒/扫描要求。9.分离原理:使用色谱棒的[color=#000000][url=http://www.runyangyiqi.com/]薄层色谱仪[/url][/color],色谱棒是外包薄层吸附剂的特殊柱子。10.执行标准:符合《SY/T 5119-2008 岩石中可溶有机物及原油族组分分析》、《SH/T 0753-2005 润滑油基础油化学族组成测定法》标准要求 。
薄层色谱法的展开时间怎么选择
芳磺酸的薄层色谱应该选什么样的展开剂呢另外我用甲醇和正庚烷体系,展开后不成斑点而是散开的一片,是不是因为两者互溶性太差的问题?有经验说芳磺酸极性太大加冰醋酸能消除拖尾,但似乎不起作用,加氨水反而拖尾现象减轻,谁能解释一下为什么呢?谢了
这篇文献是天津师范大学化学系白桦等人的文章,摘要:本文通过对实验室回收的薄层色谱展开剂的再次实验所得数据与新配制的展开剂实验所得数据进行比较,指出薄层色谱展开剂是可以再次利用,且分离效果较好。这样不仅可以节省实验经费,而且有利于环境保护,减少环境污染。
摘要:薄层色谱非常适合于药用植物的分析。由于有大量的参数可供调节以得到不同的色谱结果,薄层色谱具有其它方法所无法比拟的灵活性,这也是它所固有的优点。在另一方面,这些参数缺少标准化的精确规定,导致薄层色谱很难重现。就象现在欧洲药典中所表现的情况那样,现代薄层色谱所具有的良好特征都被广泛忽视。以下这篇文章主要对改进药典方法表述以及单个品种各论进行讨论。对实验的优化和标准化进行详细讨论,以提高方法的重现性。用单个参数的理论探讨和以一些实例来说明现代高效薄层色谱的优点以及薄层色谱方法标准化的必要。主题词:衍生化,薄层记录,高效薄层色谱,方法学,薄层展开,薄层点样,标准化,薄层色谱1、前言传统薄层色谱法被广泛应用于药用植物的分析中,并作为植物药鉴别方法被世界上多数药典收录。薄层色谱一般方法学的描述通常并不详细,并给操作者留有太多的空间。因此用这种方法所得到的结果很值得考虑,有时甚至会难以判断。由于在薄层色谱中影响结果的许多参数被忽视,薄层色谱结果的重现性常常不能令人满意。这是阻碍任何方法现代化的巨大阻力。因此,出现了这样一种论调,认为薄层色谱是一种陈旧过时的技术,应该用更可靠的高效液相色谱或其它色谱技术代替它。但是大家不应忽视薄层色谱具有众多的优点。无论在植物药的鉴别,提取物和最后产品的稳定性测试,还是在生产过程中的进程控制,薄层色谱可将结果用一个图片表现出来的这个优点不可替代。快速的分析和每个样品分析的低成本也是它的优点。一个应用于新的药典各论的现代标准方法是让大家认识和接受薄层色谱是具有竞争力的现代分析方法的基本要求。这篇文章讨论了薄层的基本要素和实际工作中的要求,达到了这个最基本的目标。希望能促进这个建设性议题的讨论。2、薄层色谱在药典中的现状在欧洲药典中,薄层色谱在所有的植物药、大多数提取物以及一些合成药物的各论中作为鉴别的基本手段。对于合成药物来说,似乎有一种趋势,在新增或修订的各论中不再使用薄层色谱作为有关物质检查的方法。虽然与其它药典相比,欧洲药典在薄层色谱的一般方法学部分具有较高的水平,但是仍没有反映在方法学方面的一些技巧以及当前技术所带来的好处。虽然规定只能使用预制薄层板,但是没有明确普通薄层板和高效薄层板之间的差别,也未说明哪种薄层板是首选。缺少薄层色谱一些重要实验细节(如薄层点样、薄层展开和衍生化)的指导性原则,甚至没有提到通过图象分析进行定性分析这一薄层最重要的优点。虽然在一般方法学部分提到薄层色谱可作为定量方法,但是只有两个品种使用薄层色谱进行定量。所有的含量测定都使用高效液相色谱,甚至在鉴别中薄层色谱也被成功地用其它技术替代。如果采用现代薄层色谱技术,可以很容易地改变这种状况。3、关于方法学改进的建议3.1薄层板的材料和特性在70年代末期,Merck研制出了高效薄层板。它具有更小的粒度(5μm)和更窄的粒度分布范围(2-10μm)。与传统薄层板相比,高效薄层板具有更好的均一性,更平整的表面和更高的分离性能。表1比较了两者的差别。(略)假设药典原来用普通薄层板的方法以高效薄层板替代。我们就可以得到具有更好分离度,更少斑点扩散以及更好的板间重现性的结果。瑞士植物化学专业委员会已经在几个方法中确认了这个设想,并在欧洲药典注释中出版。例如,图1比较了苦橘油和甜橘油在两中薄层板上的分离情况。可以得出结论高效薄层板是最好的也是最经济的选择。虽然在各论中不能规定商品的品牌,但是也应引起注意,不同的生产厂家生产的薄层板即使通过了药典试剂章节要求的系统适应性试验,也会明显地影响到某一特定方法结果所得的重现性。(图2)因为很难设计一个实验对薄层板分离可能碰到的所有问题进行评价,在进行方法描述时应对薄层板的生产厂家进行规定,或者在方法中包含一个专门的效能测定实验(系统适应性实验)。这些工作已经要求作为各论的详细技术指导在研究方法时考察并提交给药典。然而似乎并没有规定在完成各论前的方法学考察中实际包含两种材料的薄层板,在方法的确认时也没有考察不同的薄层板材料,除非这些工作已经包含在各论中。这些都应该有一个大家可以接受的法定的要求。3.2薄层点样薄层鉴别都是基于比较薄层比移值(Rf值)。因此分析的质量依赖于样品的正确定位。在定量分析中,点样体积也应规定并可重现。此外,分离质量还取决于点样原点的大小、形状以及斑点均一性。将溶液中的样品转移到薄层板上有接触式和喷雾式两种点样方法。在点状点样时,样品溶液形成“环状色谱”,导致样品在点样原点上的不均匀分布。在展开后,斑点可能展宽或不均匀。当样品溶解于对其溶解性很强的溶剂中时,这种现象更为严重。采用喷雾点样方式可显著提高薄层系统的分离度和检出限。它也可将浓度低的样品以大体积点于薄层板上,同时不影响分离的质量。样品条带状点样,便于色谱的目视。如果这些条带是通过喷雾产生的,在整个条带上的样品的均一性又可得到进一步的提高。这是得到可靠的和可重复的定量分析结果的基础。应该注意,用一个接一个的小点点样所形成的条带或用所谓的浓缩带原点并不能得到那样好的分离度,其比移值小的斑点会受到干扰。(图3B-D)3.3展开薄层色谱的结果(斑点的位置、形状和分离度)依赖于展开缸的类型和饱和程度,如图4所示。因此一个方法只在特定的展开缸中以特定的方式展开才可具有重现性,如不进行调整,在其它系统中很难得到相同的结果。相关的理论我们在其它文章中有详细说明。虽然常常使斑点较未饱和或双槽展开缸扩散大,但经典的饱和步骤(在展开缸或水平展开槽中)可获得最好的重现性。在薄层色谱的展开过程中,展开剂的移动速度不断减小(如图5)。因为采用更小的粒子使薄层板更为紧密从而阻碍了展开剂的运动,高效薄层色谱板只能展开很短的距离。在一个固定的展开槽中,固定其它参数不变,两个成分的分离度不单取决于它们在薄层色谱中的相对位置(比移值),还与溶剂前沿的移动位置(展开距离)有关。图6是两个成分在高效薄层板上的分离度与展开距离之间的关系图,其中假定选择因子(α)为1.5。分离度用公式 Rs=(α-1)(RfN)1/2(1-Rf)/4。 在高效薄层色谱中,当展开距离为5-7cm时可得到最好的分离度,在展开距离为6cm时可得到最大的分离度。在硅胶板上,大多数展开剂需要7-20min展开。在一特定的色谱分离中,比移值应在0.3-0.4最佳。应调整溶剂的比例,使主要成分的位置在此范围之间。这些理论的推测可以很容易地用实验验证。在图7中是甘菊油的高效硅胶薄层分离结果。当展开距离增加时,在图7A中比移值0.4-0.5(箭头所示)的两个斑点的分离度似乎有所增加。但是如果把这些色谱图放大到同一个刻度,两个斑点的相对位置似乎没有变化。分离度仍然在展开距离为6cm时最大。一般来说,随着展开距离的增大,比移值减小。这种现象可能是由于展开剂中的挥发性成分在薄层板上逐渐增加的所引起。同样刻度的相似曲线图也可得出与薄层图相似的结果。
[color=#444444]中药提取出来分成水溶性和脂溶性两种提取液,用薄层色谱点样看分离程度,展开剂分别如何选择?[/color]
薄层色谱所用展开剂有哪些要求?各位帮下!
薄层色谱常用的展开剂的组合有哪些?
[font=宋体]链接:[/font]https://bbs.instrument.com.cn/topic/7762216问题描述:怎么选择薄层色谱展开剂?解答:[font=宋体]薄层色谱展开剂的选择具有更大的灵活性。一般展开剂体系选择如下:根据分离样品性质、薄层色谱板性质选择一个二元溶剂体系,通过调节溶剂比例以寻求适合[/font]RF[font=宋体]值,适合的[/font]RF[font=宋体]值找到后,再寻求极性参数相同的二元溶剂体系两个,以这三个组成为三点组成一个三角形,则可看到:三角形顶点是二元体系,边是三元体系,三角形内是四元体系,并且极性一致,可根据几何原理得出任一点组成。这种方法较为直观,也较简单。[/font][font='Times New Roman','serif'][/font]以上内容来自仪器信息网《样品前处理实战宝典》
色谱分析中展开剂是如何根据物质的极性进行选择的 ?
薄层色谱分析步骤及注意事项薄层色谱法(thin layer chromatography简写TLC)是一种物理化学的分离技术,常用于药物的分离与分析。现对此方法的分析步骤及注意事项提点建议。 完成TLC分析通常需经制板、点样、展开、检出4步操作。⑴制板 在一平面支持物(通常为玻璃)上,均匀地涂制硅胶、氧化铝或其他吸附剂薄层、样品的分离、检测就在此薄层色谱板上进行。 一般选用适当规格的表面光滑平整的玻璃板。常用的薄层板规格有:10cm×20cm、5cm×20cm、20cm×20cm等。称取适量硅胶,加入0.2%~0.5%羧甲基纤维素钠溶液(CMC-Na),充分搅拌均匀,进行制板。一般来说10cm×20cm的玻璃板,3~5g硅胶/块;硅胶与羧甲基纤维素钠的比例一般为1:2~1:4。制好的玻璃板放于水平台上,注意防尘。在空气中自然干燥后,置1l0℃烘箱中烘0.5~lh,取出,放凉,并将其放于紫外光灯(254nm)下检视,薄层板应无花斑、水印,方可备用。⑵点样 用微量进样器进行点样。点样前,先用铅笔在层析上距末端lcm 处轻轻画一横线,然后用毛细管吸取样液在横线上轻轻点样,如果要重新点样,一定要等前一次点样残余的溶剂挥发后再点样,以免点样斑点过大。一般斑点直径大于2mm,不宜超过5mm.底线距基线1~2.5cm,点间距离为lcm左右,样点与玻璃边缘距离至少lcm,为防止边缘效应,可将薄层板两边刮去1~2cm,再进行点样。⑶展开 将点了样的薄层板放在盛在有展开剂的展开槽中,由于毛细管作用,展开溶剂在薄层板上缓慢前进,前进至一定距离后,取出薄层板,样品组分固移动速度不同而彼此分离。① 展开室应预饱和。为达到饱和效果,可在室中加入足够量的展开剂;或者在壁上贴两条与室一样高、宽的滤纸条,一端浸入展开剂中,密封室顶的盖。② 展开剂一般为两种以上互溶的有机溶剂,并且临用时新配为宜。③ 薄层板点样后,应待溶剂挥发完,再放人展开室中展开。④ 展开应密闭,展距一般为8~15cm。薄层板放入展开室时,展开剂不能没过样点。一般情况下,展开剂浸入薄层下端的高度不宜超过0.5cm。⑤ 展开剂每次展开后,都需要更换,不能重复使用。⑥ 展开后的薄层板用适当的方法,使溶剂挥发完全,然后进行检视。⑦ Rf值一般控制在0.3~0.8,当Rf值很大或很小时,应适当改变流动相的比例。⑷ 斑点的检出 展开后的薄层板经过干燥后,常用紫外光灯照射或用显色剂显色检出斑点。对于无色组分,在用显色剂时,显色剂喷洒要均匀,量要适度。紫外光灯的功率越大,暗室越暗,检出效果就越好。展开分离后,化合物在薄层板上的位置用比移值(Rf值)来表示。化合物斑点中心至原点的距离与溶剂前沿至原点的距离的比值就是该化合物的Rf值。
多肽的水溶性太强,薄层色谱用什么展开[color=#333333]用正丁醇-冰醋酸-水 3.5:2:4.5展开,rf只在0.1左右,且不成斑点,是一片不规则形状,似水迹阴开,是怎么回事?[/color]
若化合物本身无色,薄层色谱的色谱图中组分迁移产生的展开斑点位置如何确定?
[color=#00008B](一)有机合成中展开剂的选择 做有机合成时走板子是常有的事,展开剂的选择就至关重要了。选择适当的展开剂是首要任务.一般常用溶剂按照[/color][color=#00FFFF][color=#DC143C]极性从小到大的顺序排列大概为:石油迷己烷苯乙醚THF乙酸乙酯丙酮乙醇甲醇[/color][/color]使用单一溶剂,往往不能达到很好的分离效果,往往使用混合溶剂通常使用一个高极性和低级性溶剂组成的混合溶剂,高极性的溶剂还有增加区分度的作用,展开剂的比例要靠尝试.一般根据文献中报道的该类化合物用什么样的展开剂,就首先尝试使用该类展开剂,然后不断尝试比例,直到找到一个分离效果好的展开剂。展开剂的选择条件:①对的所需成分有良好的溶解性;②可使成分间分开;③待测组分的Rf在0.2~0.8之间,定量测定在0.3~0.5之间;④不与待测组分或吸附剂发生化学反应;⑤沸点适中,黏度较小;⑥展开后组分斑点圆且集中;⑦混合溶剂最好用新鲜配制。 一般来说,[color=#00FFFF][color=#DC143C]弱极性溶剂体系的基本两相由正己烷和水组成[/color][/color],再[color=#DC143C]根据需要加入甲醇、乙醇,乙酸乙酯来调节[/color]溶剂系统的极性,以达到好的分离效果,适合于[color=#DC143C]生物碱、黄酮、萜类等的分离[/color]; [color=#DC143C]中等极性的溶剂体系由氯仿和水基本两相组成[/color],由[color=#DC143C]甲醇、乙醇,乙酸乙酯等来调节,适合于蒽醌、香豆素,以及一些极性较大的木脂素和萜类的分离[/color]; [color=#DC143C]强极性溶剂,由正丁醇和水组成,也靠甲醇、乙醇,乙酸乙酯等来调节,适合于极性很大的生物碱类化合物的分离[/color]。 很多时候,展开剂的选择要靠自己不断变换展开剂的组成来达到最佳效果。我们在实验中,为了实现一个配体与其他杂质有效分离,曾经尝试了很多种的溶剂组合,最后才找到石油醚—EtOAc—HCOOH(5.5:3.5:0.1)混合溶剂。一般把[color=#DC143C]两种溶剂混合时,采用高极性/低极性的体积比为1/3的混合溶剂,如果有分开的迹象,再调整比例(或者加入第三种溶剂),达到最佳效果;如果没有分开的迹象(斑点较“拖”),最好是换溶剂[/color]。对于[color=#DC143C]在硅胶中这种酸性物质上易分解的物质,在展开剂里往往加一点点三乙胺,氨水,吡啶等碱性物质来中和硅胶的酸性。[/color](选择所添加的碱性物质,还必须考虑容易从产品中除去,氨水无疑是较好的选择。) 分离效果的好坏和所用硅胶和溶剂的质量很有关系:不同厂家生产的硅胶可能含水量以及颗粒的粗细程度,酸性强弱不同,从而导致产品在某个厂家的硅胶中分离效果很好,但在另一个厂家的就不行。溶剂的含水量和杂质含量对分离效果都有明显的影响。温度,湿度对分离效果影响也很明显,在实验中我们发现有时同一展开条件,上下午的Rf截然不同 。 展开剂的选择主要根据样品的极性、溶解度和吸附剂的活性等因素来考虑,在进行薄层层析时,首先应该知道未知化学成分的类型,其极性的大致归属,从提取液或从色谱柱的流动相极性可知,另外某样品里含多种化学成分先按极性不同大致分,然后细分,对于分离未知的化学物质,展开剂的选择也是一个摸索的过程,不应该仅仅从展开剂考虑,多因素综合衡量!溶剂:层析过程中溶剂的选择,对组分分离关系极大。在柱层析时所用的溶剂(单一剂或混合溶剂)习惯上称洗脱剂,用于薄层或纸层析时常称展开剂。洗脱剂的选择,须根据被分离物质与所选用的吸附剂性质这两者结合起来加以考虑在用极性吸附剂进行层析时,[color=#00008B][color=#DC143C]当被分离物质为弱极性物质,一般选用弱极性溶剂为洗脱剂;被分离物质为强极性成分,则须选用极性溶剂为洗脱剂。[/color][/color]如果对某一极性物质用吸附性较弱的吸附剂(如以硅藻土或滑石粉代替硅胶),则洗脱剂的极性亦须相应降低。在柱层操作时,被分离样品在加样时可采用于法,亦可选一适宜的溶剂将样品溶解后加入。溶解样品的溶剂应选择极性较小的,以便被分离的成分可以被吸附。然后渐增大溶剂的极性。这种极性的增大是一个十分缓慢的过程,称为“梯度洗脱”,使吸附在层析柱上的各个成分逐个被洗脱。如果极性增大过诀(梯度太大),就不能获得满意的分离。溶剂的洗脱能力,有时可以用溶剂的介电常数(ε)来表示。介电常数高,洗脱能力就大。以上的洗脱顺序仅适用于极性吸附剂,如硅胶、氧化铝。对非极性吸附剂,如活性炭,则正好与上述顺序相反,在水或亲水住溶剂中所形成的吸附作用,较在脂溶性溶剂中为强。 被分离物质的性质 被分离的物质与吸附剂,洗脱剂共同构成吸附层析中的三个要素,彼此紧密相连。在指定的吸附剂与洗脱剂的条件下,各个成分的分离情况,直接与被分离物质的结构与性质有关。对极性吸附剂而言,成分的极性大,吸附住强。当然,中草药成分的整体分子观是重要的,例如极性基团的数目愈多,被吸附的住能就会更大些,在同系物中碳原子数目少些,被吸附也会强些。总之,只要两个成分在结构上存在差别,就有可能分离,关键在于条件的选择。要根据被分离物质的性质,吸附剂的吸附强度,与溶剂的性质这三者的相互关系来考虑。首先要考虑被分离物质的极性。如被分离物质极性很小为不含氧的萜烯,或虽含氧但非极性基团,则需选用吸附性较强的吸附剂,并用弱极性溶剂如石油醚或苯进行洗脱。但多数中药成分的极性较大,则需要选择吸附性能较弱的吸附剂(一般Ⅲ~Ⅳ级)。采用的洗脱剂极性应由小到大按某一梯度递增,或可应用薄层层析以判断被分离物在某种溶剂系统中的分离情况。此外,能否获得满意的分离,还与选择的溶剂梯度有很大关系。现以实例说明吸附层析中吸附剂、洗脱剂与样品极性之间的关系。如有多组分的混合物,象植物油脂系由烷烃、烯烃、舀醇酯类、甘油三酸醋和脂肪酸等组份。如对于C-27甾体皂甙元类成分,能因其分字中羟基数目的多少而获得分离:将混合皂甙元溶于含有5%氯仿的苯中,加于氧化铝的吸附柱上,采用以下的溶剂进行梯度洗脱。如改用吸附性较弱的硅酸镁以替代氧化铝,由于硅酸镁的吸附性较弱,洗脱剂的极牲需相应降低,亦即采用苯或含5%氯仿的苯,即可将一元羟基皂甙元从吸附剂上洗脱下来。这一例子说明,同样的中草药成分在不同的吸附剂中层析时,需用不同的溶剂才能达到相同的分离效果,从而说明吸附剂、溶剂和欲分离成分三者的相互关系。 (二)簿层层析:薄层层析是一种简便、快速、微量的层析方法。一般将柱层析用的吸附剂撒布到平面如玻璃片上,形成一薄层进行层析时一即称薄层层析。其原理与柱层析基本相似。 1.薄层层析的特点:薄层层析在应用与操作方面的特点与柱层析的比较。 2.吸附剂的选择:薄层层析用的吸附剂与其选择原则和柱层析相同。主要区别在于薄层层析要求吸附剂(支持剂)的粒度更细,一般应小于250目,并要求粒度均匀。用于薄层层析的吸附剂或预制薄层一般活度不宜过高,以Ⅱ~Ⅲ级为宜。而展开距离则随薄层的粒度粗细而定,薄层粒度越细,展开距离相应缩短,一般不超过10厘米,否则可引起色谱扩散影响分离效果。 3.展开剂的选择:薄层层析,当吸附剂活度为一定值时(如Ⅱ或Ⅲ级),对多组分的样品能否获得满意的分离,决定于展开剂的选择。中草药化学成分在脂溶性成分中,大致可按其极性不同而分为无极性、弱极性、中极性与强极性。但在实际工作中,经常需要利用溶剂的极性大小,对展开剂的极性予以调整。[/color][/color][/color][/color]
薄层色谱法中,上行展开至多少cm即可,上行展开的终止位置是指展开剂的位置还是样品点到达的位置??求解
谈薄层色谱展开剂选择 根据本人的几年薄层层析经验,参考药典等国家药品标准和有关文献,将 2000版药典一部里部分有代表性的对照品的薄层层实例按展开剂极性排序,并对其规律做一些分析。以下的分析和介绍是总体描述性的,目的是快速、简便地选择展开剂。如果想了解展开剂选择的各种理论,请参考其他专著。 选择展开剂,要依据溶剂极性和他们的混溶性,溶剂对被分析物的溶解性,以及被分析物的结构。这里只讨论药典里通常使用的以硅胶为固定相主体的正相薄层,也不考虑板的活性。 列出溶剂极性参数表,方便以下比较展开剂。环已烷 :-0.2、石油醚(Ⅰ类,30~60℃)、石油醚(Ⅱ类,60~90℃)、正已烷:0.0、甲苯:2.4、二甲苯:2.5、苯:2.7、二氯甲烷:3.1、异丙醇:3.9、正丁醇:3.9、四氢呋喃:4.0、氯仿:4.1、乙醇:4.3、乙酸乙酯:4.4、甲醇:5.1、丙酮:5.1、乙腈:5.8、乙酸:6.0、水:10.2 [1] 。 关于溶剂混溶性,一般根据相似相溶原则,需要注意,极性相差大的不混溶,比如正己烷与甲醇。多元展开剂,主体的两种溶剂不能混溶,就需要通过第三种溶剂来调和。比如:石油醚、正庚烷、正已烷、戊烷、环已烷和甲醇、水之类的。 一般正相色谱,固定相为极性,被分析物质的极性越大,需要极性更大的展开剂。 了解被分析物的极性可以通过分析其结构获得,很难获得它的极性指数。物质分子化学结构中,通常由较极性部分和非极性部分两部分。例如下面以苯丙烷为极性小部分,随着极性基团部分的增加,总体的极性就增加,展开剂极性也增加了。 , 依次为肉桂酸、阿魏酸、咖啡酸、菊苣酸、绿原酸。 相应展开剂分别为:正己烷—乙醚—冰醋酸 (5:5:0.1)、苯-冰醋酸-甲醇(30:1:3)、氯仿-甲醇-甲酸(9:1: 0.5)、石油醚-乙酸乙酯-甲酸(3:6: 1)、醋酸丁酯-甲酸-水(7:2.5:2.5)。(由于薄层板、比移值不同的原因,展开剂极性比较是相对的,并非绝对的后者大于前者)。 现在最重要的问题是,不同化合物,怎么定它的极性,又用什么标准来定它对应的展开剂呢?以下分开讨论不同化合物极性情况及其对应的展开剂: 首先是极性较小的挥发性物质。比如:冰片:石油醚 (30~60℃)—醋酸乙酯(17:3)、厚朴酚:苯-醋酸乙酯(9:1.5)、α-香附酮:苯-醋酯乙酯-冰醋酸(92:5:5)、丹皮酚:环己烷-醋酸乙酯(3:1),这类化合物,以石油醚、正构烷和苯为体积百分数比较大的溶剂,通常起溶解和分离化合物的作用,而用醋酸乙酯为调节Rf(比移值)的溶剂。为了减少拖尾之类其他相似相溶原则以外的影响,适当加入添加剂,如有机酸或者有机碱。 极性较小的不挥发性物质。比如:β -谷甾醇:环己烷-醋酸乙酯-甲醇(6:2.5:1)或者环己烷-丙酮(5:2) 、熊果酸:甲苯-醋酸乙酯-冰醋酸(12:4:0.5)、齐墩果酸:氯仿-甲醇(40:1)、猪去氧胆酸:氯仿-乙醚-冰醋酸(2:2:1)、大黄素:苯—醋酸乙酯—甲醇(15:2:0.2)或者苯—乙醇 (8:1)、丹参酮ⅡA:苯-醋酸乙酯-甲酸(40:25:4) 、穿心莲内酯:氯仿-无水乙醇(9:1)、靛玉红、靛蓝氯仿-乙醇(9:1)或者苯-氯仿-丙酮(5:4:1)。这类物质展开剂极性比极性较小的挥发性物质洗脱力强一些,因为这类物质极性小的母核大,而极性大的基团通常可以形成氢键,比如羧酸、羟基。 以上物质,母核分子量减小、母核结构中不饱和健的增加(尤其是出现苯环),极性基团的增加,都使极性增加,展开剂极性也增大。这个范围内的物质很多,一般展开剂大百分数的溶剂可以从环己烷—〉甲苯—〉二甲苯—〉苯—〉氯仿的顺序,按照极性要求选择。这里注意,异丙醇、正丁醇极性指数也比较小,在这范围的化合物很少用,因为粘性大、展开慢,造成斑点扩散;另外,羟基的氢键作用力也有不利。调节Rf值的溶剂,从醋酸乙酯—〉甲醇—〉丙酮—〉乙醇。挥发性物质也有很多带羰基、羟基的,但从它的挥发性就可以明白,分子间作用力不强,另外,母核与石油醚、正构烷和苯的结构差异小,估计更容易脱离硅胶吸附,更快进入溶剂中,而不需要通过提高展开剂的极性。 皂苷类。人参皂苷:氯仿-甲醇-水 (65:35:10)10℃以下放置的下层溶液或正丁醇-醋酸乙酯-水(4:1:5)的上层溶液或氯仿-醋酸乙酯-甲醇-水(15:40:22:10)10℃以下放置的下层溶液、芍药苷:氯仿-醋酸乙酯-甲醇-甲酸(40:5:10:0.2)、黄芩苷:醋酸乙酯-丁酮-醋酸-水(10:7:5:3)、橙皮苷:苯—醋酸乙酯—甲酸—水(1:12:2.5:3)的上层溶液、葛根素:氯仿-甲醇-水(14:5:0.5)、芦丁:醋酸乙酯-甲酸-水(8:1:1)。这类物质,由于存在糖的多羟基结构,苷元的结构影响变小。展开剂中使用极性大的有机溶剂(氯仿、醋酸乙酯、甲醇、正丁醇)和水。乙酸和甲酸的使用,一方面增大展开剂极性,另外也可以抑制硅胶羟基的作用,减少拖尾。由于混溶性和硅胶耐酸能力的限制,水和酸的使用是有限度的。 极性大的小分子有机酸。没食子酸:氯仿-醋酸乙酯-甲酸 (5:4:1)、阿魏酸、咖啡酸、菊苣酸、绿原酸、异绿原酸。这类物质多数是苯乙烯母核的,这个结构的极性本身比较大,另外有酚羟基和羧酸基团,个别有多羟基配基。皂苷的展开剂差不多,极性大。注意甲酸通常指的是浓度85%左右的,含有水。 含氮有机物。盐酸小檗碱:苯-醋酸乙酯-甲醇-异丙醇-浓氨试液(12:6:3:3:0.6)(氨蒸气饱和) 或正丁醇-冰醋酸-水(7:1:2)、麻黄碱:氯仿-甲醇-浓氨试液(20:5:0.5)或正丁醇-冰醋酸-水(8:2:1)、甘草酸铵:醋酸乙酯-甲酸-冰醋酸-水(15:1:1:2)。由于NH2硅醇基的作用很强,在强极性展开剂加有机酸、有机碱扫尾。对于极性化合物,使用正丁醇对斑点扩散影响较小,因为化合物和硅胶的作用强。 进行薄层分析基本可以根据母核、基团,选择相似的化合物对号入座。当然,具体的条件优化则需要根据实际情况了。遇到较困难的分离,需要使用到设计优化方法的,已经不属于本文讨论范围了。

求助:我们实验室最近进行薄层色谱预制板展开性能方面的试验。需要使用多种成分混合染料的样品。经查询CAMAG公司的资料上有六种颜色混合染料的样品(如图)。但是国内代理公司无法提供该混合染料样品及配方资料。特求助有关专家能否提供上述或其他有关混合染料的配方,以及采用何种相应的展开剂。谢谢!http://ng1.17img.cn/bbsfiles/images/2017/04/201704231919_01_1783468_3.jpg







 我要推广仪器
我要推广仪器
 下载APP
下载APP