色谱柱的湿法和干法填柱,有哪些区别?
请问大家一下,农残分析中干法装柱和湿法装柱的区别是什么呀?不同的吸附剂用哪种方法好呀?有什么优缺点吗?新手求助,谢谢啦![em0904]
干法就是把待分离的样品用少量溶剂溶解后,在加入少量硅胶,拌匀后再旋去溶剂。如此得到的粉末再小心加到柱子的顶层。干法上样较麻烦,但可以保证样品层很平整。湿法上样就是用少量溶剂(最好就是展开剂,如果展开剂的溶解度不好,则可以用一极性较大的溶剂,但必须少量)将样品溶解后,再用胶头滴管转移得到的溶液,沿着层析柱内壁均匀加入。然后用少量溶剂洗涤后,再加入。湿法较方便,熟手一般采用此法。
[b]【序号】:1【作者】:[b]李天玉,张宇航[/b]【题名】:干法UO2粉末松装密度和振实密度的影响因素研究【期刊】:中国核科学技术进展报告(第六卷)——中国核学会2019年学术年会论文集第6册【年、卷、期、起止页码】:[font=&][size=12px][color=#333333]130-137[/color][/size][/font]【全文链接】:https://kns.cnki.net/kcms/detail/detail.aspx?dbcode=CPFD&dbname=CPFDLAST2020&filename=EGVD201908006021&uniplatform=NZKPT&v=yJX-OjiJJTcjJecE9tSgfEri1w9hEefX8ko9Pt_1qzrJ35Pl4di6b9JbGbgGlx7pcLmkEIKVab0%3d[/b]
学校实验室现有色谱装机的项目,欲购色谱装柱机一台,不知道国内哪家做的好。
请教各位,装色谱柱要注意哪些问题?如涂布、装柱的气压、老化等问题?谢了!
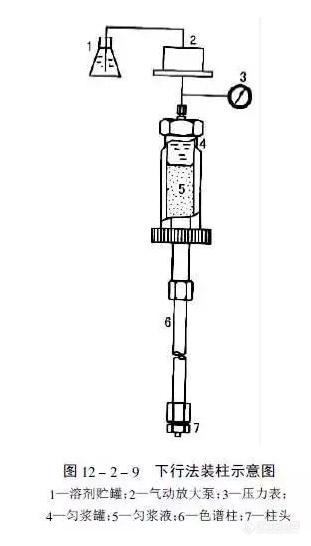
[align=center][b]色谱柱是如何填装的?[/b][/align]目前大多数实验室是购买商品预填装柱来满足分离、分析之需。液相色谱柱、特别是高效液相色谱柱的填装,需要有较高的技巧和熟练的技能。因此,有人甚至将“装柱”看作是“艺术加技术”。在有关色谱基本理论的讨论中可以得知,发生在色谱柱中总的谱带展宽效应与流动相的线速度、粒径以及溶质在流动相中的扩散系数、溶质在固定相中的扩散系数等密切相关。对于给定粒径的填料来说,能否填充成均匀而紧密的柱床,是得到高性能柱子的关键,而采用粒径细且分布均匀的优质填料,则是得到高性能柱子的最基本保证。[b]高压匀浆法装填[/b]将填料悬浮在适宜的匀浆液中制成匀浆,在其尚未沉降之前,很快以高压泵将其以很高 的流速压进柱中,便可制备出填充均匀的柱子。这是常见的分析和制备色谱柱的装填方法。常用的“标准”HPLC柱为φ4.6mm×250mm,其内腔体积约为4.2mL,约需3.5g填料。[img=,311,546]https://ng1.17img.cn/bbsfiles/images/2018/12/201812131515258469_1462_2428063_3.png!w311x546.jpg[/img][b]有关分析色谱柱的思考我认为自己装填色谱柱是有风险的,商品化色谱柱的装填是熟练技术工人千锤百炼的结果有自己独特和专用的装填设备,塞板的选择和放置都是非常精确地。如果装填技术不过关,在高压作用下,填料透过塞板,进入检测器是非常危险的,还有可能损坏检测器。所以尽可能的选择商品化的色谱柱本身是有质量保证的!本文部分节选于化工信息网微信,经作者整理加工而成[/b]
[em02] 请问有哪位高人知道色谱柱的装法?借点知识.谢谢![em53]
大家好:请问装柱的技巧是什么?怎样装才不能花柱子?
制备色谱柱根据固定相颗粒度和柱子的尺寸,采用不同的装柱方法,往往装填越好分离效果越好。装柱效果跟填料的颗粒度关系很大,颗粒度的减少会导致装柱的难度。一般来说,颗粒直径小于20-30um的固定相采用湿法装填。所谓“敲击-装填”技术适用于颗粒直径大于25um的固定相。湿法的目的是迫使相对稀松的固定相悬浆以高速装入色谱柱子,从而减少空隙的形成。然而,当柱直径大于20mm,所加压力为30-40bar时,高压悬浆装填技术就变得十分复杂。为将小颗粒固定相装入更大得制备型色谱柱,可采用柱长压缩技术。这种方法,先将固定相悬浆(或偶尔是干填充物)装入柱中加压,利用物理方法将其压紧。压紧的方法有两种:径向压缩和轴向压缩。湿法装柱需要一定的设备,在柱子填完后,应用有柱效的测量,对柱效低的柱子应该重填。
海德利森色谱装柱流程以填装5um 硅胶 4.6×250 分析柱为例 1 连接装柱所需空压机和海德利森色谱装柱机 确保装柱机工作时机身稳固2 称取填料3.0-3.5g (4.6×250 分析柱标准需要2.8g,经验3.2g比较合适)3 用40-50ml异丙醇(或异丙醇与乙腈比例1:1)溶解填料 用玻璃棒搅拌均匀 后用超声波超3分钟 以确保其分散性4 迅速将上述混合液倒入匀浆罐(尽量短时间内完成该工序并确保匀浆罐内无气泡) 如匀浆罐不满再用异丙醇(或异丙醇与乙腈比例1:1)补满 理想情况是不需要再补充异丙醇(或异丙醇与乙腈比例1:1)熟练后可知需要多少原料 所需原料的梁与温度、填料溶解性有关5 迅速将匀浆罐和柱子连接到泵上 以60MPA的压力 以甲醇(化学纯)做流动相开启装柱机 保持装柱机在60MPA压力下工作20分钟6 减压至30MPA 运行5-10分钟 缓慢降压至系统无压力 静止3分钟7 卸下柱子 看柱头部分是否平整 如不平整用美工刀铲平(很需要技术的步骤)8 将柱子接入高效液相色谱仪 用甲醇(色谱级)以1.0流速运行30分钟9 开始检验色谱柱效10 多次熟练此工作 以提高装柱质量备注:1异丙醇(或异丙醇与乙腈比例1:1)至少分析纯2甲醇 为控制成本可用化学纯 但事先需要经过0.2um 有机滤膜过滤3工作开始前开启装柱机用流动相甲醇冲洗管路 尽量确保管路中无气泡 北京海德利森科技有限公司. 陈海涛 工程师 13911876986 cht01310751@163.com
今天不小心把一根液相色谱柱(C18)装反了,跑了一个上午,还进两个样,中午的时候发现装反了,就赶紧换回来了(这有可能是最致命的),还回来之后出峰的形状变差了,到晚上的时候色谱峰就开始分叉了。现在不知道该怎么办了,不知道还有没有补救措施啊?http://simg.instrument.com.cn/bbs/images/default/em09508.gif
我现在的实验室还没有条件使用微波消解仪,所以我们在测镉时都是采用的湿法消解和干法灰化,在消解的过程中我发现干法灰化对不同的样品最终灰化结果具有较大的差异,所以我想将这些贴出来让大家讨论一下,请各位老师前辈指点。方法:1、样品有两种——生鱼胚(海水鱼小鱼干)、成品鱼(生鱼胚经过泡水、油炸、拌料等过程制成);2、向瓷坩埚中称取样品1g左右,在电炉子上碳化至无烟;3、将碳化好的样品放入马弗炉中,500摄氏度7小时;4、向取出冷却后的瓷坩埚中加入混酸(硝酸:高氯酸=9:1)5mL,在电炉上加热消解至透明或淡黄色,溶液渐干时取下冷却硝酸溶液溶解。疑惑:1、在做的过程中我发现样品坩埚放在马弗炉炉腔不同的位置灰化效果不同,可以通过怎样的方法能够平衡;2、成品鱼在最后加混酸消化烤至渐干后,溶解的时候会出现一些不溶物,类似透明状的小石子,而生鱼胚从未出现这种情况;3、国标中说样品中灰化至浅灰色或白色时可以直接溶解上机测,这对仪器是否会有影响?
我在做食品中矿物质及重金属地测定时用干法和湿法处理样品做对照,发现对像钠、钾这样地元素,两种方法产生地误差奇大,接近10倍。而锌误差也在3倍左右。其他地元素地情况在后面继续补充。备注:干法:在电热板120度烧至无烟,转移入马弗炉600度烧近4h。取出冷却后用稀硝酸或稀盐酸溶解。测定。湿法:用硝酸和高氯酸4:1约20毫升120度电热板加热消解至无色或淡黄色,赶酸,用二次水或稀酸定容。测定。锌:湿法测试结果是干法地三分之一。钾:湿法较干法高10%左右。钠:湿法是干法地10倍。各位大侠有遇到过这样地结果吗?谢谢!!!
如题请问哪位大侠使用装柱机装填过液相色谱柱?哪个型号的装柱机比较好用?使用装柱机装填液相色谱柱有哪些技巧及需要注意的地方?谢谢了:)
在化学检验中,消解处理是为了排除有机物和悬浮物干扰,将各种价态的元素氧化成单一高价态或转变成易于分离的无机化合物,从而得到清澈透明无沉淀的浓缩溶液用于检测分析;消解处理主要应用于钙、镁、钠、锌、铜、铁、磷等微量成分的分析。 消解方法主要有: 干法消解(灼烧法)、湿法消解、微波消解。1.干法消解(灼烧法) 干法消解是通过高温碳化、灰化除去大量有机物,然后用酸或其它溶剂溶解,制成试样溶液,最后主要用溶剂萃取、掩蔽、沉淀等方法排除其它离子的干扰。 优点:①因加入试剂少,故空白值低;②因灰分体积小,可处理较多的样品,可富集被测组分;③有机物分解彻底,操作简单。缺点:①所需时间长,温度高易造成易挥发元素的损失;②坩埚对被测组分有吸附作用,使测定结果和回收率降低。 操作注意事项: ①灰化前样品应进行预炭化,其中炭化、加硝酸溶解残渣等操作必须在通风橱内进行; ②不宜使用新的瓷坩埚进行灰化,避免新瓷坩埚吸附待测成分,造成实验误差; ③若样品灰化不完全(主要表现为颗粒状或黑色),可将坩埚取出,冷却后,加入少量硝酸或水湿润残渣,干燥后再移入高温炉内继续灰化; ④从高温炉中取出坩埚时,应先放置于炉口冷却,避免高温灼伤;切忌直接置于木制台面、有机合成台面上以免烫坏台面,也不宜直接置于导热系数较高的台面上,以免徒然遇冷引起坩埚破裂。
色谱柱装树脂吸废水中的金属,色谱柱起了什么作用呢?
制备装柱机,分析装柱机,色谱装柱机,国内哪家做的比较好?想要进口的,
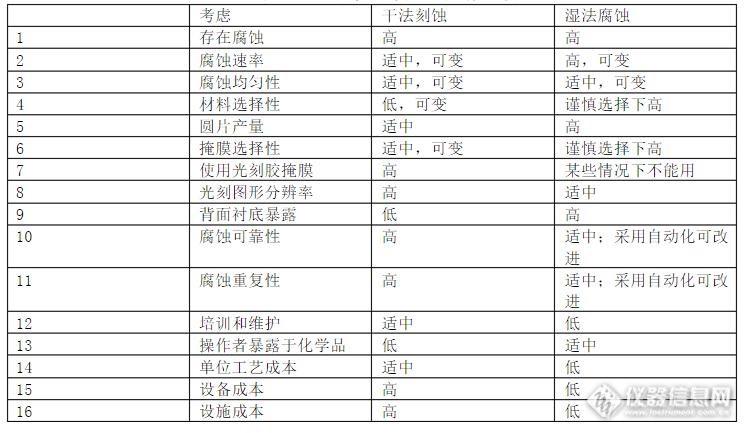
湿法腐蚀是使用液态腐蚀剂系统化的有目的性的移除材料,在光刻掩膜涂覆后(一个曝光和显影过的光刻胶)或者一个硬掩膜(一个光刻过的抗腐蚀材料)后紧接该步腐蚀。这个腐蚀步骤之后,通常采用去离子水漂洗和随后的掩膜材料的移除工艺。http://ng1.17img.cn/bbsfiles/images/2016/12/201612130959_01_3091062_3.jpg干法刻蚀的刻蚀剂是等离子体,是利用等离子体和表面薄膜反应,形成挥发性物质,或直接轰击薄膜表面使之被腐蚀的工艺。http://www.whchip.com/upload/201612/1481592347583553.jpg湿法腐蚀可替换工艺包括干法刻蚀,即使用一种或多种低压力的反应气体,采用RF感应激励后进行反应,然后再将反应生成的气态物质抽出;非等离子干法刻蚀,例如双氟化疝或氢氟酸的酸性蒸气腐蚀,拥有各向同性湿法腐蚀的诸多特性,该腐蚀通常在一个有限的腔室内完成。很少有微机械化或集成化的器件是在没有进行一些湿法化学处理的情况下开发或制造的。不管器件是否是电气的,机械的,电子的,集成的,光学的,光电子学的,生物的,聚合的,微流控的传感器或执行器,有关这些器件的制造工艺或过程的替换决定将对最终的技术和商业成功有重要影响。这些器件通常在硅衬底、化合物半导体、玻璃、石英、陶瓷或塑性材料上制造,可能涉及在这些材料上淀积一层或多层薄膜并光刻和腐蚀。这些层和淀积顺序受工艺和用于开发和制造该器件的工艺单元限制,随着层数的增长变的越来越复杂和相互影响。 近乎所有IC,MEMS,MOEMS,MST和NEMS类的器件的产生都很可能与一些湿法腐蚀工艺有关。整个工艺流程可被描述为一系列步骤或者序列,这些湿法腐蚀常用于选择性的去除淀积薄膜的一部分,剥去诸如硬掩膜和光刻胶等特定的材料,为以后的加工清洗和准备衬底,去除牺牲层和部分衬底,以及形成三维结构。一个湿法腐蚀工序需要考虑如下一些因素,包括有效的腐蚀剂,腐蚀选择性,腐蚀速率,各向同性腐蚀,材料的兼容性,工艺的兼容性,花费,设备的可用性,操作人员的安全,技术支持和适当的废物处理。干法刻蚀能实现各向异性刻蚀,保证细小图形转移后的高保真性。器件设计者,工艺设计师,或者制造商在工艺允许的情况下可能偏向使用一个完整的干法处理流程,但是许多标准的处理步骤例如光刻胶的显影和圆片清洗仍然湿法的。与干法刻蚀相比,湿法腐蚀工序在成本,速度,性能发面更有优势。干法刻蚀的仿真还不可用,如常用的微结构的选择性钻蚀或与晶向相关的腐蚀仿真等。考虑到干法刻蚀要求在一个昂贵的等离子区或者RIE腐蚀系统里有长的腐蚀时间,湿法腐蚀变得特别有吸引力,需要同时处理整盒圆片(25片装圆片盒)或更多的圆片时,湿法腐蚀在成本和时间上的效益更突出。 不管选择干法还是湿法加工工艺,总是强烈受到在特定的加工环境下设备的可用性及对开发者有用的工艺限制。成功的设计者,开发者和制造商几乎总是使用或修改趁手的工艺。除非是必须开发新工艺,安装新设备,或者取得新的工艺技能,一般总是避免额外的需求。理解什么时候要应用干法和湿法这两个工艺并且在可能的情况下使用标准工艺是很重要的。下表总结比较湿法和干法刻蚀之间的一般注意事项。 http://ng1.17img.cn/bbsfiles/images/2016/12/201612130959_02_3091062_3.jpg
专用色谱、装柱机、柱温箱、及各种色谱耗材供应 代理MERCK公司一系列产品联系电话:010-84923350 010-84923351联系人:苗先生
本人看到,哈希电导率手册上有干法校准,什么是干法校准?电导率如何进行干法校准?弄不明白啊!!!!!
干法处理对坩埚的要求请问给位前辈,干法灰化处理样品对坩埚的材质有没有要求呢?石英、瓷还是铂坩埚?希望能回复详细点,谢谢。
我的色谱图峰形状呈现后拖尾,而且骑着侧峰,请教诸位会是什么问题?原来峰形很好很对称的,标准品出现的问题,我觉得是色谱柱的问题,需要清洗或者衍生,诸位给点意见啊?

[table=100%][tr][td]现在买了一台北京海德利森的标准型色谱装柱机框架式,但是还不会使用,产品型号:HY-HPLC-S,我们现在除了一台机器以外什么都没有,所以我想知道1、除了这台仪器还需要什么仪器或者部件(匀浆管的接口与成品色谱柱不配套,连接匀浆管和色谱柱一定还需要什么部件)2、可不可以给出一份详细的使用步骤(包括各个仪器的使用顺序,试剂的添加顺序等),拿具体的物质举例就可以,因为我们现在还不确定要填装什么,只是希望有一个流程附带装柱机的图片,有知道的希望能不吝赐教,谢谢了[/td][/tr][/table][img=,690,516]https://ng1.17img.cn/bbsfiles/images/2019/09/201909161009096913_2524_1827556_3.jpg!w690x516.jpg[/img]
液相色谱柱是如何填装的?
液相色谱柱是如何填装的
请问热导检测器是否必须装两根色谱柱?
请各位大虾帮忙说一下液相色谱柱的填装的最佳方法!
有几个问题想请教一下我是新手,在做铅工作曲线的时候有时候有不良点,回归偏差比较大,在这种情况下,可否将其删除,而保留好的点还有就是,我们处理样品的时候(做铅),现在用湿法,还要赶酸,(先在电炉上,再上水浴),做起来很麻烦。用干法的回收率太低,而且极不稳定,回收率甚至出现负数。请问用干法处理时有没有什么防止铅损失的措施!
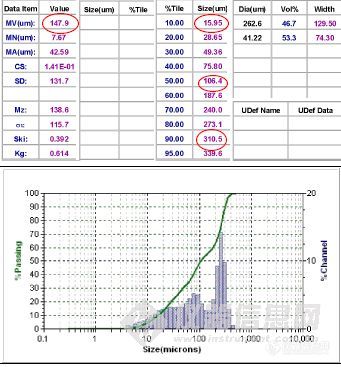
[size=3]本人需要测试自己实验室合成的聚酰亚胺粉末的粒度,之前采用马尔文激光粒度仪(干法)测试过,因为不懂,不知道还有湿法,看论坛讲湿法测试的重复性更好,请问各位经验人士,我们这样的聚合物用湿法合适吗?不溶于水和乙醇,但在水和乙醇中是沉淀状态,不是悬浮状态[/size][img]http://ng1.17img.cn/bbsfiles/images/2010/05/201005051512_216480_1667949_3.jpg[/img]







 我要推广仪器
我要推广仪器
 下载APP
下载APP