用的中压柱,内径22mm,长50cm,耐压10bar,填料是sephadex LH20,流动相是甲醇和水,梯度是100%水,20%甲醇,40%甲醇,60%甲醇,80%甲醇,100%甲醇。开始100%水冲柱子的时候,压力大约在4bar左右,但是随着甲醇增加,梯度逐渐变大,最后100%甲醇时候,超过了15bar,柱子开始漏液了。开始我流速设置的0.5ml/min,最后压力到了11bar,后来我把流速改到了0.3ml/min,最后压力反而超过了15bar。这究竟是怎么回事?上样之前我用各个恒梯度都试过的,压力都不超过8bar,谁知道一跑样就超了。
岛津2010A,纯甲醇时,柱压8.6Mpa,甲醇:水(10:90)时,柱压15.2Mpa,乙腈:水(85:15)时柱压7.5Mpa,乙腈:水(82:18)时,柱压15.3Mpa,柱压是否过高?可能是什么原因造成的?应该如何解决?本人对仪器不太懂,求助各位专家,感谢各位????
现有一台GC112A色谱,柱前压1.2公斤,桥电流190mA,汽化室温度柱室温度检测室温度测乙酸乙酯含量时,甲醇不限水要求小于11%,乙醇小于3%,甲酯小于3%,水保留时间0.75分,乙醇为1.25分,但水拖尾严重,水和乙醇分不开请问老师应该怎样调试,才能完全分开?
[align=center][size=18px]减压阀和取样针法对高纯气中氧氮的影响[/size][/align][align=left][size=16px]高纯气体或电子气体分析中,通常采用的方式是检测气体中杂质气的含量,100%减去气体中杂质含量的总和得到气体的纯度,氧气和氮气是多数气体分析中的杂质,精确分析氧气和氮气对纯度的影响非常关键。[/size][/align][align=left][size=16px]怎样精确的取到原气体,气体样品不在导入分析仪器时出现失真非常关键。对于高压气体通常用的取样方式及取样工具有减压阀和取样针法,这两种取样方式各有优缺点。[/size][/align][align=left]1, [size=16px]减压阀,[/size][size=16px]从结构看,减压的效果非常好,可以从几十兆帕的压力减压到公斤级,而且减压的稳定性也非常好,对于气体分析中的取样,缺点是有一定的死体积,特别是痕量的氧气和氮气杂质非常难吹扫干净:气体分析离线分析时需要快速的吹扫取样设备和仪器系统,假设按开一次瓶阀再关一次瓶阀,放空余压为一次吹扫,曾做过实验,用减压阀操作100次以后,残留的氧气和氮气还会剩余1ppm以下[/size][size=16px],耗时大概30小时左右[/size][size=16px]。[/size][/align][align=left]2, [size=16px]取样[/size][size=16px]针阀,从稳压效果看,远远赶不上减压阀,但是从结构看吹扫起来特备容易,同样[/size][size=16px]按开一次瓶阀再关一次瓶阀,放空余压为一次吹扫,曾做过实验,用减压阀操作[/size][size=16px]8[/size][size=16px]次以后[/size][size=16px],基本吹扫干净,耗时3分钟不到。[/size][/align][align=left][size=16px]综上,对于气体分析,特别是同一台仪器分析不同基体中杂质的含量时,取样针法吹扫的速度远远超过减压阀的速度。[/size][/align]
新手,第一次做.看见2487关机前还要清洗,这个清洗会经过柱子吗?我用的是C18的柱子,据说不能过100%纯水?那我怎么清洗2487?另外,现在在走流动相,甲醇:已腈=3:7,要看基线是不是平稳,是不是也要通过监视器看,监视器上能看见红线在浮动,但是我的2487上的吸广度是ERROR,是怎么回事呢?新问题: 出现上面问题后,我重起系统,就没有ERROR了.柱压大概是900左右,用的是millunnium32的软件。开了监视器,走了一会流动相后。就在“单一”那个窗口,按“进样”的那个纽后出现等待,监视器就自己关掉了。然后进样,把进样的阀打到LOAD,进样后,阀打到inject,能听到“滴”的一声。但是m32的那个窗口所有选项一直是灰的,只有进样那个纽可以用,而且一直是waiting状态。监视器也是灰的。然后点“查看”都没有线出来。请问是怎么回事?晚上甲醇洗一遍后(30分钟多)就关机了第二天过来,直接走的甲醇:已腈=3:7,结果就发现柱压很不稳定,非常不稳定。而且最高可以升到2000多。又是怎么回事啊??急!![em06] 上面的问题都解决了.还有一个就是氧化甾醇用什么做溶剂更好?我用甲醇,而且用0.2um的有机滤膜滤过,结果就把柱子堵了。
要分析样品中的乙醇、乙醚、乙醛,用什么色谱柱呀?如果样品中还有乙烯、、环氧乙烷,能用一根柱子分开吗?该用什么柱子?谢谢!
[size=5]最近学校买了台天津博纳艾杰尔agela CHEETAHTM系列中压快速纯化制备系统,以前没用过此类制备色谱,请问哪问有CHEETAHTM使用说明啊?请多多指教![/size]
一根waters c18新柱,用它分析一种黏性很强的物质,在4.5mins出峰,USP tailing 1.09 但最近发现柱压2500 psi,比刚开始用时高了400 psi,出峰时间不变,主峰USP tailing 0.90,变成了前沿峰(在新配了流动相后,以前也有换过,但拖尾不变),流动相为 甲醇:水:三乙胺:磷酸。 请问:原因何在?是不是柱子损害比较严重?
前辈们,我这一台安捷伦液相1120型八月份才换的柱子。这几天柱压升高到150几。用甲醇洗了好几个小时,柱压还是降不下来。这么办跪求方法?柱子C18
压汞仪中的纯汞(规格99.999999)大家是哪里买的?
甲醇的柱压怎么是乙腈的两倍啊?我用乙腈/水=70/30,1ml/min,柱压46bar,但用甲醇/水=60/40,1ml/min,柱压90bar,是这样的吗?今天第一次换甲醇/水,柱压呼呼呼的上升,吓我一跳,不知道换流动相中间需要怎么过渡?我直接用配好的甲醇/水流动相就冲柱子了
各位XDJM,在空分分析系统中,氩中氮分析仪(0-10ppm)零点标定都是用的什么纯化器呢?最近要采购一台,要求知名进口品牌,麻烦大家推荐下,谢谢。
色谱柱在使用中最常见的问题就是柱压升高,如果柱压是在长时间使用过程中缓慢增加,属于正常现象。但柱压在使用过程中突然升高(系统管路堵塞及压力传感器故障除外),以下列举了部分常见原因及解决办法: (1)色谱柱头的过滤筛板堵塞或污染; 解决方法:如确定是色谱柱头的过滤筛板被污染,可以将色谱柱反方向用甲醇冲洗至正常压力,或者卸下色谱柱头,将其放在10%的稀硝酸内超声清洗10分钟,后再用纯水超声10分钟,重新装入色谱柱。 (2)色谱柱头的填料被样品污染; 解决方法:如确定色谱柱头的填料被污染,将柱头螺丝卸下,挖出柱内前段被污染的填料,用相同的柱填料重新填入,仔细修复后,重新安装上柱头螺丝。 (3)色谱柱内缓冲液中的盐遇到高浓度的甲醇或其他有机溶剂,形成结晶析出; 解决方法:如确定定是盐结晶,用10%的甲醇/水冲洗色谱柱使柱内盐全部溶解,再换高浓度甲醇。 (4)生物样品分析导致的柱压升高 解决方法:0.1%三氟乙酸水溶液冲洗色谱柱。若10倍柱体积冲洗后压力不降,则无法恢复。 (5)流动相PH值过大或过小使固定相结构破坏或溶解。 解决方法:如果因PH值使用不当,很难恢复。
单位的一台液相色谱,在使用纯甲醇做流动相在设定方法下分析一种样品的时候,柱压会增大到200以上,同样的方法分析同样的样品,换了一台仪器不存在这个问题。两台仪器都是使用C18柱子,但是不是一根,出峰时间很近,几乎没有差别。再次想请教各位老师专家,出现这种情况,可能是仪器哪里出现了问题,过滤头,滤芯都已经换了。
在用异丙醇冲柱子时的柱压一般在多少?柱子:3微米、150mm、流速:0.2
高纯氩中微量氮测定,除了气相色谱法和专用的仪器测定,有没有其他方法可以测定啊?
各位大侠怎么用MS测高纯金中的钌、铱、钨、钛

铸铁平板平台压砂前的预备工作:研磨方法 铸铁平板平台的研磨方法一般有两种:一种是三板互研法,这种方法压砂的结果是,三块平板平面度都很好,三块平板的压砂效果基本一样,并且三块平板都可以使用,不用常常压砂,但对修理技术要求较高。所谓三板互研法是指三块平板相互之间依次互研,并且每块平板只能当下板两遍,实际共研磨6遍。 另一种方法是两块板互研法,也叫子母板压法。这种方法是只用两块平板一上一下互研,用这种方法压砂结果是,两块平板的平面度基本吻合,上面平板的平面度凹,下板的平面度凸,并且下板的压砂效果要比上板的好。因为上板中间凹,不轻易修理量块,一般不用上板,只用下板。缺点是下板的突出程度不易把握,只能用一块平板。每次压砂时需要研磨3~4遍才可以完成,每遍6分钟左右。两种方法比拟较,本人经验觉得仍是前者较好。平板的选用 压砂前的预备工作很重要。首先选用什麽样的铸铁平板平台?采用那种压砂方法?选用什麽规格的砂子?心里应该清晰。修理量块的平板一般有灰铸铁、高磷球墨铸铁、高磷低金属球墨铸铁等三种。平板的硬度在HB (130~250)之间。采用硬度较低的平板压砂,嵌粗砂轻易,适合于粗研,修出的量块表面粗拙度比较低,表面发白。采用硬度较高的平板,嵌粗砂难题,轻易嵌入细砂,耐磨性不理想,修出量块的表面粗拙度高,光彩青亮。从我二十来年的工作经验看,采用硬度在HB(180~210)的平板,既压砂轻易,耐用,又合用于高精度的精密研磨,特别合用于量块修理。 金刚砂规格的选用 平板的压砂有两种方法:(1)从开始到结束只用一种规格的砂子,修理量块一般用M2.5规格的砂子.(2)压砂从开始到结束,砂子从粗到细循序渐进地压砂,一般选取M4,M3,M2.5规格的砂子。 研磨平板需要的辅料 (1)混合油:煤油和变压器油的比例3:1。 (2)硬脂。 (3)用汽油泡好的砂子即金刚砂。 平板恒温 修理室的温度很重要,太高或太低都不轻易嵌砂,一般温度在(20±5)°C。有的单位压砂房间恒温前提比较差,当平板从一温度拿到另一温度的房间研磨时,平板受热涨冷缩现象的影响,表面产生变形,故等温一段时间,一般平板在压砂前等温数小时或更长时间,待平板变形不乱后,再开始研磨。 平板的修整 对于新铸铁平板平台和表面划痕、碰伤较重的平板,先用油石打磨一下,打磨后,我们先用M4的砂子修一下板面,三块平板互相研磨,待推拉费力的时候,卸下平板,直到平板的平面度较好时,休止修理平板,做到冷暖自知。 对于使用中划痕、碰伤较轻的平板,只用油石把突起的部门打磨下去就行,不用修理平板,直接压砂就可以了铸铁平板平台的压砂 从我们多年来的工作经验看,选用(300×300)mm的平板,采用三板互研法压出来的平板修理量块最为相宜,由于它简朴易操纵,只用一种规格的砂子,不用多次换砂,并且压一次砂三块平板都可以使用。 首先我们把第一块平板放在工作台上,用汽油把平板擦干净。在铸铁平板平台上涂上少许硬脂,然后倒上用汽油泡好的M2.5的金刚砂(一吸管的量),等汽油挥发后,滴入 10滴混合油,把下板涂匀,再在上板的四角及中央处涂些,这样可以防止在压砂开始时,因为油膜厚度不均两板之间形成一个楔角,会泛起“啃”板现象。 前两次压砂所达到的推拉力和所用油量一样,后四次压砂滴入7~8滴混合油,推拉力达到750N左右,这时的推拉速度达每行程10秒左右。 每遍压砂大约需要20分钟左右。把握了以上压砂要领,就可以达到预期的效果。我们用这种方法得到的研磨平板用试块试,手感:⑴很柔和;⑵切削力很快。目测试块:切削痕迹平均即粗拙度好。这样的平板经久耐用,可以修理量块250~300块,我们一年只需要压砂3~4次,就可以保证全市各大企事业单位、计量站的量块修理量。
请教各位大侠高人:我用的是黛安的液相,以前用的是国产的甲醇,但最近走甲醇时柱压突然就很高,以前是30左右,现在60而且有上升到趋势,请各位老师指点迷津,是什么原因造成的?是柱子堵了么?我用异丙醇冲了效果也不明显,该如何补救?请各位大侠帮忙!!!!!!!!!!是柱前的单项阀不畅所致???如果是,该怎么拆卸冲洗呢??能给发个单向阀的实图么?
请教各位大侠高人:我用的是黛安的液相,以前用的是国产的甲醇,但最近走甲醇时柱压突然就很高,以前是30左右,现在60而且有上升到趋势,请各位老师指点迷津,是什么原因造成的?是柱子堵了么?我用异丙醇冲了效果也不明显,该如何补救?请各位大侠帮忙!!!!!!!!!!是柱前的单项阀不畅所致???如果是,该怎么拆卸冲洗呢??能给发个单向阀的实图么?
苯基柱走纯盐走了一晚上后,用10%甲醇,0.5的流速冲洗,未接检测器,柱压越来越高怎么办
各位大哥大姐: 听说好多柱子都不能用纯水走,还是有一些可以走的,各位高手你们能够告知一下哪些色谱柱可以用纯水走呀?我想归纳一下,谢谢大家了。
岛津LC-20A ,泵无压力,检测发现柱头出有点漏液,拧紧了,未漏液,但泵的压力还是没有!A泵(甲醇)无压力,泵管内有一段气泡B泵(纯水)有压力我运行了一段,只有B泵有压力,A泵无,是不是说明只有B泵的水进入柱子了,A泵的甲醇根本没进入柱子,那不是等于我用纯水冲柱子了吗? 急救啊 我的柱子会不会完蛋了!怎么补救呀!泵没有压力,又怎么办呢?请高手帮忙呀!http://simg.instrument.com.cn/bbs/images/brow/em09509.gif
实验室想对高纯氩中杂质分析。气体情况如下:高纯氩:氦≤4ppm、氧≤1.5ppm 氢≤5ppm 甲烷≤1ppm 水≤3ppm不知道版友有没有做过或者接触过类似情况的项目的。多谢。
4.6*250 5um的C18柱 1ml/min的甲醇下 压力大概多少 柱温室温 大概15度左右我不接检测器有75kgf+ 现在反冲中。。。。昨天结束后用异丙醇冲洗,早上回来发现0.3ml/min下压力有180+[em0810] 一段段看下来好像柱子压力有点高 咨询下 我不接柱的压力有6-7kgf 仪器是岛津LC10-AT的
前两天测雄果酸样品,流动相为甲醇:0.2%乙酸铵(85:15),流速1ml/min,柱压最大30MPa。 熊果酸标品以甲醇做溶剂,样品提取试剂也是甲醇。开始用流动相平衡的时候,柱压为15MPa左右。因为是新柱子,新仪器,平衡的时间长一点,平衡及跑标样过程中,柱压逐渐升高到18MPa,最后一次做样品时,柱压升高到20MPa。清洗柱子的时候,考虑到流动相中盐浓度较高,可能会有乙酸铵晶体堵塞柱子。开始一直用纯水冲洗,但柱压高,0.5ml/min时,柱压能升到22MPa以上,用甲醇:水(85:15),0.5ml/min冲洗时,柱压反而能降到17MPa左右,后来改用纯甲醇或纯水冲洗,流速不变,柱压都在20MPa以上.流速升高到1ml/min柱压就会超过30MPa。今天尝试以甲醇:乙腈(80:20),流速1ml/min冲洗,柱压很快降到13MPa,并维持稳定,后来换成纯甲醇,柱压保持在15-16MPa。但换成纯水冲洗,0.5ml/min时,柱压还是在22MPa以上,流速1ml/min时柱压又会超过30MPa。 实在是不得其解,请高手指点该如何将柱子冲洗干净。忘了说了,柱子是rainbow C18反相柱4.6*250 mm。
问题:甲醇中的七种苯系物怎么分开呀。感觉苯和甲醇分不开p,用FFAp,DBWAX,都试过方法:因为你试的都是极性柱子,极性柱子分不开,非极性柱子可分开。甲醇在极性柱子会拖尾,所以你买二硫化碳作溶剂的苯系物就可以了。(盲样就是甲醇中的七种苯系物)。那就两个仪器跑,用极性柱做一条曲线,再用非极性做一条曲线。用你的-5柱子去做苯的,就没问题,其他也可以的,只不过对间是合在一起的。 如果盲样允许对间一起合报,那抹黑就直接用非极性了。看看说明,基本上都允许合报的,专家有给你们一张结果报告要求,里面就有说允不允许。大家怎么看?
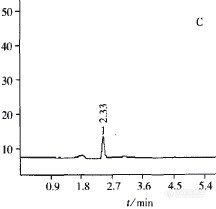
液相色谱法测定水体中的亚硝态氮、硝态氮和总氮N02-、NO3-及总氮已列入水体的必检项目,建立水体中N02-、NO3-及总氮的新型、快速、准确检测方法显得极其重要。目前,同时测定亚硝酸盐和硝酸盐的方法主要采用化学发光法、硝酸银电极法、流动注射光度法、二元线性解析一紫外分光光度法及离子色谱法等。这些方法各有优缺点,其中离子色谱法已经被我国列入生活饮用水中N02-和NO3-检验规范方法之一。离子色谱法的采用,改善了N02-和NO3-检测方法的灵敏度,但不普及。1 实验部分1.1仪器与试剂安捷伦HPLC1200。亚硝酸钠、硝酸钾、磷酸、氢氧化钠和过二硫酸钾均为分析纯;磷酸二氢钾为pH缓冲标准物质;乙腈为色谱纯;100 mg/L N02-标准样品、500 mg/L NO3-一N总氮标准样品、(12.275 ±1.55)mg/L TN标准控制样品,均为国家环保总局标样所标样。1.2 色谱条件月旭色谱柱,Ultimate XB-C18 4.6 x250,5um(part Number 00201-31043 Serial Number 211302350 Lot Number 2101.77)。柱温为30℃,流速0.8 mL/min,进样体积10ul;流动相:17.5 mmol/L KH2PO4一2 mmol/L H3P04缓冲液(pH 3.5)与乙腈体积比为92.5:7.5,使用前过滤并超声脱气。1.3实验方法每100 mL水样加1 mL缓冲液至酸性,过月旭 Welchrom Syringe Filter Nylon 13mm 0.45um(Part Number 00802-02202 Lot Number 140108)滤膜,直接进样10uL测定N02-、NO3-;按照水质总氮测定分析方法处理水样,用HPLC法测定。采集相同水样,按上述GB 11894—89消煮法消解水样后,比色法测定总氮;比色分析水样中NO2一N、NO3--N时,按GB/T 5750—2001《生活饮用水卫生规范》处理,若水样浑浊或色度较深,在100 mL水样中加2 mL氢氧化铝悬浮液,搅拌后静置数分钟,过滤。分取水样或处理后的水样用酸或碱调节至中性,用重氮偶合分光光度法测定NO2--N;另外分取水样或处理后的水样加适量硫酸银一硫酸溶液(10 g/L),混匀,静置5 min,滴加高锰酸钾溶液至淡红色保持15min不褪色后,用麝香草酚分光光度法测定N03--N。2结果与讨论2[font=宋
还是SCR-101H柱的问题:奇怪的是一开始用甲醇冲仪器柱压正常,换成氯仿后也正常;再换甲醇时,柱压又急剧上升,直到现在不能使用。
专家好!我有一根SCR-101H色谱柱,一开始用甲醇冲洗柱压正常,换成40%甲醇水溶液柱压急剧上升,直至仪器保护,为什么?







 我要推广仪器
我要推广仪器
 下载APP
下载APP